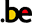
Alle versies voor 171496
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Afin de
pouvoir bénéficier d’une intervention de l’assurance obligatoire pour les
prestations relatives aux neurostimulateurs et accessoires pour stimulation
cérébrale profonde (Deep Brain Stimulation ou DBS) en cas d’épilepsie
réfractaire, il doit être satisfait aux conditions suivantes :
1.
Critères concernant l’établissement hospitalier
Les
prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566,
171570-171581, 171592-171603, 171636-171640, 171651-171662, 171673-171684,
171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780,
171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de
l’assurance obligatoire que si elles sont effectuées dans un établissement
hospitalier qui répond aux critères suivants:
1.1. L’établissement
hospitalier est inscrit sur la liste des centres spécialisés pour l’épilepsie
réfractaire (centres de référence – 7893)
1.2. L’établissement
hospitalier dispose d’une équipe multidisciplinaire responsable de la pose
d’indication, de l’évaluation pré-chirurgicale, de l’implantation, y compris
les remplacements, de la rééducation et du suivi à long terme et qui est
composée au moins d’un épileptologue, d’un neurochirurgien et d’un psychiatre.
L’épileptologue
est un médecin-spécialiste ayant une expérience démontrée et maintenue dans le
domaine du traitement de l’épilepsie tel
que défini dans la convention de rééducation avec des centres de référence pour
bénéficiaires souffrant d’épilepsie réfractaire.
L’établissement
hospitalier doit garantir une permanence
en neurochirurgie et en neurologie 24 heures sur 24 et 7 jours sur 7.
Le suivi
du bénéficiaire au quotidien est permis hors de ces centres de référence.
2.
Critères concernant le bénéficiaire
Les
prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566,
171570-171581, 171592-171603, 171636-171640, 171651-171662, 171673-171684,
171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780,
171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de
l’assurance obligatoire que si le bénéficiaire répond aux critères suivants :
2.1.
Critères d’inclusion
1) Le
bénéficiaire est atteint d’épilepsie focale avec des crises focales complexes,
avec ou sans généralisation secondaire.
2) Le
bénéficiaire est atteint d´épilepsie réfractaire, c´est-à-dire qu’un contrôle
satisfaisant des crises ne peut pas être obtenu avec un des médicaments
antiépileptiques potentiellement efficaces, administré seul ou en combinaison,
à des doses thérapeutiques optimales et non associées à des effets secondaires
inacceptables, les crises entrainant des incapacités et un handicap.
3) Le
bénéficiaire doit avoir été traité par un traitement pharmacologique optimal.
Le bénéficiaire n’a pas de contrôle satisfaisant de l’épilepsie avec au minimum
3 thérapies différentes, dont au minimum une association, aux doses optimales et durant une période
suffisante pour en apprécier l’efficacité.
4)
L’indication et la pertinence d’un traitement par stimulation cérébrale
profonde par rapport à une chirurgie de l’épilepsie, une stimulation du nerf
vague ou d’autres alternatives telles que définies dans les revues
systématiques de la littérature publiées les plus récentes, ont été discutées
et évaluées, préalablement à l’intervention, par l’équipe multidisciplinaire.
5) Le
bénéficiaire n’est pas éligible pour une chirurgie ou la chirurgie de
l’épilepsie est un échec. L’évaluation préchirurgicale doit être réalisée et
inclut les tests suivants :
a. Enregistrement vidéo-EEG de longue
durée avec enregistrement des crises
b. IRM à haute résolution du cerveau
c. FDG-PET du cerveau
d. Évaluation neuropsychologique incluant
les éléments suivants :
i. QI
ii. Mémoire
iii. Fonctions exécutives frontales
e. Évaluation psychiatrique incluant entre
autres les éléments suivants :
i. Inventaire de dépression de Beck
ii. QoLIE-31
Au cas où
un de ces examens ne serait pas réalisable, par exemple suite à un retard
mental (qui ne constitue pas en soi une contre-indication à l´implantation d´un
stimulateur pour stimulation cérébrale profonde), la raison doit être
clairement mentionnée dans le compte-rendu de la concertation
multidisciplinaire.
6) Si le
bénéficiaire est traité par stimulation du nerf vague (VNS) et qu’un contrôle
satisfaisant des crises n’a pas pu être obtenu, la stimulation VNS doit avoir
débuter au minimum 2 ans avant l’implantation d’un neurostimulateur DBS.
7) Le
bénéficiaire doit être âgé d’au moins 18 ans au moment de l’implantation.
8) Le
bénéficiaire (ou éventuellement son responsable légal) est capable d’utiliser
un programmateur et de se soumettre aux tests demandés.
9) Seuls
les bénéficiaires (ou éventuellement leur responsable légal) clairement
capables, de décider de leur plein gré, via une déclaration de consentement
éclairé, de l’implantation d’électrodes et d’un neurostimulateur entrent en
ligne de compte. Cette déclaration doit expliquer de manière détaillée les
avantages et inconvénients du traitement, les risques ainsi que l’impact
psychosocial.
2.2.
Critères d’exclusion
1)
Affection neurologique ou médicale grave qui constitue une contre-indication
pour une intervention cérébrale.
2) Toute
contre-indication chirurgicale pour subir une DBS, y compris les
contre-indications connues pour la DBS et/ou pour l’exécution d’une IRM
préopératoire, contre-indications dans le cadre d’une intervention sous
anesthésie ou autres facteurs à risque pour une intervention chirurgicale (une
affection cardio-vasculaire grave, coagulopathie,…).
3)
Utilisation inappropriée d’un produit (alcool, drogue,…), abus de substance qui
ne permet pas un usage correct de l’appareil ou rendant un suivi
médical/psychiatrique systématique impossible.
4) Idées
suicidaires.
5)
Problématique chronique psychotique non stabilisée sous traitement, à
l’exception de la psychose péri-ictale.
3.
Critères concernant le dispositif
Les
prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566,
171570-171581, 171592-171603, 171636-171640, 171651-171662, 171673-171684,
171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780,
171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de
l’assurance obligatoire que si le dispositif répond aux critères suivants:
3.1.
Définition
Le
neurostimulateur est un générateur d’impulsions électriques équipé d’une
batterie, l’ensemble étant totalement implanté chez le bénéficiaire. Le
neurostimulateur doit être relié physiquement à une ou plusieurs électrodes, et
si nécessaire au moyen d’une ou plusieurs extensions.
Le
programmateur patient est un appareil physique comprenant toutes les
applications digitales appropriées.
Les
dispositifs visés par les prestations 171636-171640 et 171651-171662 ne portent
pas le marquage CE pour l’indication d’épilepsie réfractaire mais doivent faire
l'objet d'une dérogation accordée par le Ministre ayant la Santé publique dans
ses attributions. Ces dispositifs doivent être notifiés et avoir un marquage CE
pour un traitement par neurostimulation dans une autre indication.
3.2.
Critères
Pas
d’application.
3.3.
Conditions de garantie
Afin de
pouvoir être repris sur la liste nominative pour les prestations 171496-171500,
171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603,
171791-171802 et 171813-171824, le dispositif doit répondre aux conditions de
garantie suivantes :
-
Neurostimulateurs non-rechargeables :
une
garantie totale de 24 mois doit être donnée pour les neurostimulateurs
non-rechargeables.
-
Neurostimulateurs rechargeables :
une
garantie de neuf ans doit être donnée pour les neurostimulateurs rechargeables
: une garantie totale pour les cinq premières années et une garantie au prorata
pour les quatre années suivantes. Pour le chargeur 171791-171802 et
171813-171824, une garantie totale de neuf ans est exigée. Les prestations
171636-171640 et 171651-171662 doivent également répondre à ces conditions de
garantie.
4.
Procédure de demande et formulaires
4.1.
Première implantation
Pour les
prestations 171496-171500, 171555-171566, 171673-171684, 171710-171721,
171754-171765 et 171791-171802 : Pas d’obligation administrative.
4.2.
Remplacement
4.2.1. Pour les prestations
171511-171522, 171570-171581, 171695-171706, 171732-171743, 171776-171780 et
171813-171824 : Pas d’obligation administrative.
4.2.2. La
prestation 171636-171640 ne peut faire l’objet d’une intervention de
l’assurance obligatoire qu’après accord du Collège des médecins-directeurs,
avant l’implantation, sur base du formulaire B-Form-I-21 introduit par
l’épileptologue faisant partie de l’équipe multidisciplinaire.
Le
Collège des médecins-directeurs communique sa décision motivée au pharmacien
hospitalier, au bénéficiaire concerné via son organisme assureur et à
l’épileptologue qui a introduit la demande, endéans les 30 jours suivant la
réception de la demande.
4.3.
Remplacement anticipé
4.3.1. Une intervention de
l’assurance obligatoire pour la prestation 171533-171544 ou 171592-171603 en
cas de remplacement anticipé pendant la période de garantie décrite au point
3.3., peut être accordée par le médecin-conseil après évaluation que le dispositif
remplacé ne tombe pas dans les conditions de garantie.
La
prestation 171533-171544 ou 171592-171603 ne peut faire l’objet d’une
intervention de l’assurance obligatoire qu’après accord du médecin-conseil,
avant l’implantation, demandé sur base du formulaire B-Form-I-20 entièrement
complété et signé par l’épileptologue faisant partie de l’équipe
multidisciplinaire.
Pendant
la période de garantie et en cas de dysfonctionnement qui n'est pas lié à la
pathologie du bénéficiaire ou à l'évolution de sa situation médicale, le
distributeur est obligé d'appliquer les conditions de garantie et de fournir
une note de crédit, quel que soit le distributeur qui fournit le
neurostimulateur de remplacement.
4.3.2. Pour la prestation
171651-171662, la procédure décrite au point 4.2.2 doit être appliquée.
4.4.
Dérogation à la procédure
Pas
d’application.
5.
Règles d’attestation
5.1.
Règles de cumul et de non-cumul
Une
intervention de l'assurance obligatoire pour la prestation 171496-171500 ou
171555-171566 exclut, pendant une période de deux ans, une intervention de
l'assurance pour la prestation 170892-170903.
5.2.
Autres règles
L’intervention
de l’assurance obligatoire pour la prestation 171511-171522 ne peut être
accordée que minimum deux ans après la prestation 171496-171500, 171511-171522
ou 171533-171544.
L’intervention
de l’assurance obligatoire pour les prestations 171570-171581 et 171636-171640
ne peut être accordée que minimum neuf ans après la prestation 171555-171566,
171570-171581, 171592-171603,
171636-171640 ou 171651-171662.
L’intervention
de l’assurance obligatoire pour la prestation 171813-171824 ne peut être
accordée que minimum neuf ans après la prestation 171791-171802.
5.3.
Dérogation aux règles d’attestation
Pas
d’application.
6.
Résultats et statistiques
Pas
d’application.
7.
Traitement des données
Les
données enregistrées dans le cadre de la condition de remboursement B-§09 sont
celles déterminées dans les formulaires mentionnés aux points 4.2.2. et 4.3. et
conformément aux données reprises à l'article 35septies/9 de la loi.
Le
traitement des données visées au premier alinéa s'effectue conformément aux
finalités précisées à l'article 35septies/8, 1° de la loi.
Le
traitement des données personnelles est effectué tel que mentionné à l’art. 35
septies/10, 1° et 2° de la loi.
Seules
les personnes telles que mentionnées à l’article 35 septies/11, 2° et 4° de la
loi ont accès aux données à caractère personnel non pseudonymisées.
Le délai
de conservation des données visé à l'article 35septies/13, alinéa 1er de la loi
est fixé à 10 ans.
8.
Divers
Pas
d’application.
Teneinde een tegemoetkoming van de verplichte
verzekering te kunnen genieten voor de verstrekkingen betreffende de
neurostimulatoren en hun toebehoren voor diepe hersenstimulatie (Deep Brain
Stimulation of DBS) in geval van refractaire epilepsie moet aan volgende
voorwaarden worden voldaan:
1. Criteria betreffende de verplegingsinrichting
De verstrekkingen 171496-171500, 171511-171522,
171533-171544, 171555-171566, 171570-171581, 171592-171603, 171636-171640,
171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743,
171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in
aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien
ze zijn uitgevoerd in een verplegingsinrichting die aan de volgende criteria
voldoet:
1.1. De
verplegingsinrichting staat op de lijst van de
gespecialiseerde centra voor refractaire epilepsie (referentiecentra -
7893).
1.2. De
verplegingsinrichting beschikt over een multidisciplinair team dat
verantwoordelijk is voor de indicatiestelling, de prechirurgische evaluatie, de
implantatie, met inbegrip van de vervangingen, de revalidatie en een follow-up
op lange termijn en dat is samengesteld uit minstens een epileptoloog, een
neurochirurg en een psychiater.
De epileptoloog is een arts-specialist met een
aangetoonde en onderhouden expertise op
het gebied van de behandeling van epilepsie zoals gedefinieerd in de
revalidatieovereenkomst met referentiecentra voor rechthebbenden lijdend aan
refractaire epilepsie.
De verplegingsinrichting moet een permanentie in
neurochirurgie en neurologie garanderen van 24 uur op 24 en van 7 dagen op 7
dagen.
De dagelijkse follow-up van de rechthebbende mag
buiten die referentiecentra worden verricht.
2. Criteria betreffende de rechthebbende
De verstrekkingen 171496-171500, 171511-171522,
171533-171544, 171555-171566, 171570-171581, 171592-171603, 171636-171640,
171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743,
171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in
aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien
de rechthebbende aan de volgende criteria voldoet:
2.1. Inclusiecriteria
1) De rechthebbende lijdt aan focale epilepsie met
focale complexe aanvallen met of zonder secundaire generalisering.
2) De rechthebbende lijdt aan refractaire epilepsie,
d.w.z. waarbij de aanvallen onvoldoende onder controle kunnen worden gehouden
met één van de potentieel doeltreffende
anti-epileptica, die alleen of in combinatie worden toegediend, met optimale
therapeutische dosissen waaraan geen onaanvaardbare bijwerkingen zijn
verbonden; de aanvallen kunnen bovendien arbeidsongeschiktheid of een handicap
tot gevolg hebben.
3) De rechthebbende moet een optimale farmacologische
behandeling hebben gekregen. De rechthebbende is niet in staat om de epilepsie
voldoende onder controle te houden met minstens 3 verschillende therapieën,
waarvan minstens één combinatie, met optimale dosissen en gedurende een periode
die volstaat om de doeltreffendheid ervan te beoordelen.
4) De indicatie en de pertinentie van een behandeling
door diepe hersenstimulatie ten opzichte van epilepsiechirurgie, stimulatie van
de nervus vagus of andere alternatieven zoals gedefinieerd in recentst
gepubliceerde systematische literatuurreviews werden besproken en geëvalueerd
voorafgaand aan de ingreep door het multidisciplinair team.
5) De rechthebbende komt niet in aanmerking voor
chirurgie of de epilepsiechirurgie is een mislukking. De prechirurgische
evaluatie moet uitgevoerd worden en omvat de volgende tests:
a. Langdurige
EEG-videomonitoring met registratie van de aanvallen
b. Hoge
resolutie-MRI van de hersenen
c. FDG-PET-scan
van de hersenen
d. Neuropsychologische
evaluatie die de volgende elementen bevat:
i. IQ
ii. Geheugenfuncties
iii. Frontale
executieve functies
e. Psychiatrische
evaluatie die onder andere de volgende elementen bevat:
i. Beck
Depression Inventory
ii. QOLIE-31
Indien één van deze onderzoeken niet uitvoerbaar is,
bijvoorbeeld als gevolg van een mentale achterstand (die op zich geen
contra-indicatie is voor de implantatie van een stimulator voor diepe
hersenstimulatie), dient de reden duidelijk te worden vermeld in het verslag
van het multidisciplinair overleg.
6) Als de rechthebbende met stimulatie van de nervus
vagus (VNS) wordt behandeld en er kon geen bevredigende aanvalscontrole worden
bereikt, moet VNS van start gegaan zijn minstens 2 jaar vóór de implantatie van
een DBS-neurostimulator.
7) De rechthebbende is op het ogenblik van de
implantatie minimaal 18 jaar oud.
8) De rechthebbende (of eventueel zijn wettelijke
vertegenwoordiger) kan een
patiëntenprogrameerapparaat gebruiken en is bereid om zich aan de gevraagde
tests te onderwerpen.
9) Alleen de rechthebbenden (of eventueel hun
wettelijke vertegenwoordiger) die duidelijk in staat zijn om, uit eigen wil,
via een informed consent over de implantatie van elektroden en een
neurostimulator te beslissen, komen in aanmerking. In die verklaring moeten
omstandig de voor- en nadelen van de behandeling, de risico’s en de
psychosociale impact worden toegelicht.
2.2. Exclusiecriteria
1) Ernstige neurologische of medische aandoening die
een contra-indicatie is voor een cerebrale ingreep.
2) Elke chirurgische contra-indicatie om DBS te
ondergaan, met inbegrip van de contra-indicaties die voor de DBS en/of voor de
uitvoering van een preoperatieve MRI bekend zijn, contra-indicaties in het
kader van een ingreep onder anesthesie of andere risicofactoren voor een
chirurgische ingreep (een ernstige cardiovasculaire aandoening, coagulopathie,
…).
3) Ongepast gebruik van een product (alcohol, drugs,
…)/middelenmisbruik waardoor het toestel niet correct kan worden gebruikt of
waardoor er geen systematische medische/psychiatrische follow-up mogelijk is.
4) Suïcidale gedachten.
5) Chronische psychotische problematiek die niet
gestabiliseerd is onder behandeling, met uitzondering van een peri-ictale
psychose.
3. Criteria betreffende het hulpmiddel
De verstrekkingen 171496-171500, 171511-171522,
171533-171544, 171555-171566, 171570-171581, 171592-171603, 171636-171640,
171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743,
171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in
aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien
het hulpmiddel aan de volgende criteria voldoet:
3.1. Definitie
De neurostimulator is een generator van elektrische
impulsen uitgerust met een batterij, die als geheel wordt ingeplant bij de
rechthebbende. De neurostimulator moet fysiek verbonden zijn met één of
meerdere elektroden, indien nodig door middel van één of meerdere extensies.
Het patiëntenprogrammeerapparaat is een fysiek
apparaat met alle bijhorende digitale toepassingen.
De hulpmiddelen bedoeld onder de verstrekkingen
171636-171640 en 171651-171662 dragen geen CE-markering voor de indicatie
refractaire epilepsie, maar moeten het voorwerp uitmaken van een derogatie
toegekend door de Minister die bevoegd is voor Volksgezondheid. Deze
hulpmiddelen moeten zijn genotificeerd en een CE-markering hebben voor
neurostimulatiebehandeling bij een andere indicatie.
3.2. Criteria
Niet van toepassing.
3.3. Garantievoorwaarden
Om te kunnen worden opgenomen op de nominatieve lijst
voor de verstrekkingen 171496-171500, 171511-171522, 171533-171544,
171555-171566, 171570-171581, 171592-171603, 171791-171802 en 171813-171824
moet het hulpmiddel beantwoorden aan de volgende garantievoorwaarden:
- Niet heroplaadbare neurostimulatoren:
een totale garantie van 24 maanden moet worden gegeven
voor de niet heroplaadbare neurostimulatoren.
- Heroplaadbare neurostimulatoren:
een garantie van negen jaar moet worden gegeven voor
de heroplaadbare neurostimulatoren: een volledige garantie voor de eerste vijf
jaar en voor de volgende vier jaar een garantie pro rata. Voor de lader
171791-171802 en 171813-171824 is een volledige garantie van negen jaar
vereist. De verstrekkingen 171636-171640 en 171651-171662 moeten ook aan deze
garantievoorwaarden voldoen.
4. Aanvraagprocedure en formulieren
4.1. Eerste implantatie
Voor de verstrekkingen 171496-171500, 171555-171566,
171673-171684, 171710-171721, 171754-171765 en 171791-171802: Geen
administratieve verplichting.
4.2. Vervanging
4.2.1. Voor de
verstrekkingen 171511-171522, 171570-171581, 171695-171706, 171732-171743,
171776-171780 en 171813-171824 : Geen administratieve verplichting.
4.2.2. De verstrekking
171636-171640 kan enkel in aanmerking komen voor een tegemoetkoming van de
verplichte verzekering na akkoord van het College van artsen-directeurs, voor
implantatie, op basis van het formulier B-Form-I-21, ingediend door de
epileptoloog die deel uitmaakt van het multidisciplinair team.
Het College van artsen-directeurs deelt zijn
gemotiveerde beslissing mee aan de ziekenhuisapotheker, aan de betrokken
rechthebbende via zijn verzekeringsinstelling en aan de epileptoloog die de
aanvraag heeft ingediend, binnen de 30 dagen na ontvangst van de aanvraag.
4.3. Voortijdige vervanging
4.3.1. Een
tegemoetkoming van de verplichte verzekering voor de verstrekking 171533-171544
of 171592-171603 voor een voortijdige vervanging binnen de garantietermijn
zoals bepaald in punt 3.3., kan door de adviserend-arts worden verleend na
evaluatie of het te vervangen hulpmiddel niet binnen de garantievoorwaarden
valt.
De verstrekking 171533-171544 of 171592-171603 kan
enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering
na akkoord van de adviserend-arts, voorafgaand aan implantatie, aangevraagd op
basis van formulier B-Form-I-20, volledig ingevuld en ondertekend door de
epileptoloog die deel uitmaakt van het multidisciplinair team.
Gedurende de garantieperiode en in geval van
disfunctie die niet te wijten is aan de pathologie van de rechthebbende of de
evolutie van zijn medische toestand, is de verdeler verplicht de
garantievoorwaarden na te leven en een kredietnota af te leveren, ongeacht
welke verdeler de vervangingsneurostimulator levert.
4.3.2. Voor de
verstrekking 171651-171662 dient de procedure beschreven onder punt 4.2.2
toegepast te worden.
4.4. Derogatie aan de procedure
Niet van toepassing.
5. Regels voor attestering
5.1. Cumul en non-cumulregels
Een tegemoetkoming van de verplichte verzekering voor
de verstrekking 171496-171500 of 171555-171566 sluit tijdens een periode van
twee jaar een tegemoetkoming van de verzekering voor de verstrekking
170892-170903 uit.
5.2. Andere regels
De tegemoetkoming van de verplichte verzekering voor
de verstrekking 171511-171522 kan pas minstens twee jaar na de verstrekking
171496-171500, 171511-171522 of 171533-171544 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor
de verstrekkingen 171570-171581 en 171636-171640 kan pas minstens negen jaar na
de verstrekking 171555-171566, 171570-171581, 171592-171603, 171636-171640 of
171651-171662 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor
de verstrekking 171813-171824 kan pas minstens negen jaar na de verstrekking
171791-171802 worden toegekend.
5.3. Derogatie van de attesteringsregels
Niet van toepassing.
6. Resultaten en statistieken
Niet van toepassing.
7. Verwerking van gegevens
De gegevens die in het kader van de
vergoedingsvoorwaarde B-§09 worden geregistreerd zijn deze bepaald in de
formulieren vermeld onder punten 4.2.2. en 4.3. en in overeenstemming met de
gegevens vermeld onder artikel 35septies/9 van de wet.
De verwerking van de in het eerste lid bedoelde
gegevens gebeurt volgens de doeleinden bepaald in artikel 35septies/8, 1° van
de wet.
De verwerking van de persoonsgegevens gebeurt zoals
vermeld in artikel 35septies/10, 1° en 2° van de wet.
Enkel de personen zoals vermeld in artikel 35
septies/11, 2° en 4° van de wet hebben toegang tot de niet-gepseudonimiseerde
persoonsgegevens.
De bewaringstermijn van de gegevens bedoeld in artikel
35septies/13, eerste lid van de wet wordt vastgesteld op 10 jaar.
8. Allerlei
Niet van toepassing.
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Afin de pouvoir bénéficier d’une intervention temporaire de l’assurance obligatoire pour les prestations relatives aux neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire, il doit être satisfait aux conditions suivantes :
1. But de la convention
Cette convention a pour but de fixer le remboursement temporaire de l’assurance obligatoire soins de santé ainsi que ses modalités concernant les neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire dans le cadre d’une application clinique limitée pendant la période d’évaluation qui cours du 1er décembre 2020 au 30 novembre 2023 inclus. Pendant cette période, le dispositif sera évalué selon les dispositions prévues au point 9.
2. Critères concernant l’établissement hospitalier
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si elles sont effectuées dans un établissement hospitalier qui répond aux critères suivants et qui a conclu la convention B-ACL-001-bis avec le Comité de l’assurance:
L’établissement hospitalier doit durant la totalité de la durée de la convention répondre aux critères ci-dessous.
2.1. Seuls les établissements hospitaliers ayant signé la convention de rééducation des centres de référence pour bénéficiaires souffrant d’épilepsie rebelle peuvent adhérer à cette convention.
L’établissement hospitalier doit avoir une permanence en neurochirurgie et en neurologie 24 heures sur 24 et 7 jours sur 7.
La pose d’indication, l’évaluation pré-chirurgicale, l’implantation, y compris les remplacements, la rééducation et un suivi à long terme ne peuvent être effectuées que par ces seuls établissements hospitaliers. Le Comité de l'assurance soins de santé dresse, sur proposition du Service des soins de santé, une liste, mise à jour de façon continue, avec la composition de l'équipe par établissement hospitalier et l'envoie pour information à la Commission.
L’établissement hospitalier et les médecins ayant adhéré à la convention B-ACL-001-bis s’engagent à collaborer à l’évaluation visée au point 9.
Le suivi du bénéficiaire au quotidien est permis hors de ces centres de référence.
2.2. Formulaire de candidature pour l’établissement hospitalier
L’établissement hospitalier doit se faire connaître auprès du Service des Soins de santé sur base du formulaire B-Form-II-02
2.3. Accord de coopération
L’établissement hospitalier ne peut conclure ni d’accord de coopération formalisé avec d’autres établissements hospitaliers ni d’associations hospitalières. De plus, l’intervention doit être réalisée dans les murs de l’établissement hospitalier habilité.
3. Critères concernant le bénéficiaire
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le bénéficiaire répond aux critères suivants :
3.1. Critères d’inclusion
1) Le bénéficiaire est atteint d’épilepsie focale avec des crises focales complexes, avec ou sans généralisation secondaire.
2) Le bénéficiaire est atteint d´épilepsie réfractaire, c´est-à-dire qu’un contrôle satisfaisant des crises ne peut pas être obtenu avec un des médicaments antiépileptiques potentiellement efficaces, administré seul ou en combinaison, à des doses thérapeutiques optimales et non associées à des effets secondaires inacceptables, les crises entrainant des incapacités et un handicap.
3) Le bénéficiaire doit avoir été traité par un traitement pharmacologique optimal. Le bénéficiaire n’a pas de contrôle satisfaisant de l’épilepsie avec au minimum 3 thérapies différentes, dont au minimum une association, aux doses optimales et durant une période suffisante pour en apprécier l’efficacité.
4) Le bénéficiaire n’est pas éligible pour une chirurgie ou la chirurgie de l’épilepsie est un échec. L’évaluation préchirurgicale inclut les tests suivants :
a. Enregistrement vidéo-EEG de longue durée avec enregistrement des crises
b. IRM à haute résolution du cerveau
c. FDG-PET du cerveau
d. Évaluation neuropsychologique incluant les éléments suivants :
i. QI
ii. mémoire
iii. fonctions exécutives frontales
e. Évaluation psychiatrique incluant entre autres les éléments suivants :
i. inventaire de dépression de Beck
ii. QoLIE-31
Au cas où un examen ne serait pas réalisable, par exemple suite à un retard mental (qui ne constitue pas en soi une contre-indication à l´implantation d´un stimulateur pour stimulation cérébrale profonde), la raison doit en être clairement mentionnée dans le formulaire.
5) Le bénéficiaire doit être âgé d’au moins 18 ans et de maximum 65 ans lors de l’implantation.
6) Le bénéficiaire est capable d’utiliser un programmateur et de se soumettre aux tests demandés
7) Seuls les bénéficiaires clairement capables, de décider de leur plein gré, via une déclaration de consentement éclairé, de l’implantation d’électrodes et d’un neurostimulateur entrent en ligne de compte. Cette déclaration doit expliquer de manière détaillée les avantages et inconvénients du traitement, les risques ainsi que l’impact psychosocial.
8) Si le bénéficiaire est traité par stimulation du nerf vague et en cas de résultats insuffisants, il faut minimum 2 ans depuis la primo-implantation pour pouvoir implanter un neurostimulateur DBS et inversement.
9) La zone stimulée (cible) est celle qui est reprise dans les indications couvertes par le marquage CE en vigueur.
3.2. Critères d’exclusion
1) Affection neurologique ou médicale grave qui constitue une contre-indication pour une intervention cérébrale.
2) Toute contre-indication chirurgicale pour subir une DBS, y compris les contre-indications connues pour la DBS et/ou pour l’exécution d’une IRM préopératoire, contre-indications dans le cadre d’une intervention sous anesthésie ou autres facteurs à risque pour une intervention chirurgicale (une affection cardio-vasculaire grave, coagulopathie,…).
3) Utilisation inappropriée d’un produit (alcool, drogue,…), abus de substance qui ne permet pas un usage correct de l’appareil ou rendant un suivi médical/psychiatrique systématique impossible.
4) Idées suicidaires
5) Problématique chronique psychotique non stabilisée sous traitement, à l’exception de la psychose péri-ictale
4. Critères concernant le dispositif
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le dispositif répond aux critères suivants:
4.1. Définition
Les dispositifs visés par les prestations 171614-171625, 171636-171640 et 171651-171662 ne portent pas le marquage CE mais doivent faire l'objet d'une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
4.2. Critères
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 et 171695-171706, le dispositif doit répondre aux critères suivants :
Le dispositif doit avoir fait preuve de son efficacité et sa sécurité à l’aide d’ études cliniques. Ces études seront publiées dans une revue « peer-reviewed » reconnue internationalement. Parmi les études cliniques réalisées, il faut au minimum une étude de suivi sur au minimum 100 patients suivis pendant minimum 2 ans, ainsi qu’une étude clinique comparative, randomisée au minimum pendant 3 mois et ayant une puissance statistique déterminée a priori d’au moins 80%. L’étude de suivi et l’étude randomisée peuvent être la même étude.
4.3. Conditions de garantie
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 et 171813-171824, le dispositif doit répondre aux conditions de garantie suivantes :
- neurostimulateurs non-rechargeables :
une garantie totale de 24 mois doit être donnée pour les neurostimulateurs non-rechargeables
- neurostimulateurs rechargeables:
une garantie de neuf ans doit être donnée pour les neurostimulateurs rechargeables : une garantie totale pour les cinq premières années et pour les quatre années suivantes une garantie au prorata. Pour le chargeur 171791-171802 et 171813-171824, une garantie totale de neuf ans est exigée. Les prestations 171614-171625, 171636-171640 et 171651-171662 doivent également répondre à ces conditions de garantie.
5. Nombre de bénéficiaires
Le nombre de bénéficiaires qui peuvent entrer en ligne de compte pour une intervention de l’assurance obligatoire est limité à 25 nouveaux bénéficiaires par an.
Dès que le nombre menace d’être dépassé, le Secrétariat en informe la Commission et il est demandé à l’évaluateur de communiquer un rapport. La nature du rapport est établie par la Commission.
La Commission informe les établissements hospitaliers et les distributeurs du dispositif concerné et prend les mesures nécessaires.
6. Procédure de demande et formulaires
6.1 Première implantation
La demande d’intervention de l’assurance obligatoire pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 se déroule comme suit:
La demande d’intervention pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 est introduite avant implantation par l’épileptologue sur base du formulaire B-Form-I-07 au Collège des médecins-directeurs.
Ces prestations ne peuvent faire l’objet d’une intervention de l’assurance qu’après accord du Collège des médecins-directeurs. En cas de doute, le Collège des médecins-directeurs peut transmettre la demande pour avis à la Commission Peer Review, composée des prestataires ayant signé la convention de rééducation « centres de référence pour bénéficiaires souffrant d’épilepsie réfractaire ». Cette Commission Peer Review est alors tenue d’examiner la demande de l’équipe du centre de référence dans les trois mois. La Commission Peer Review avertit l’équipe du centre de référence qui a introduit la demande afin de lui permettre de la défendre. Au cours de la discussion portant sur les dossiers qui entrent en ligne de compte pour une intervention de l’assurance pour la neurostimulation, un membre du Collège des médecins-directeurs ou un médecin, membre de la Commission, peut toujours être présent. Minimum deux membres de la Commission Peer Review issus de deux établissements autres que le centre de référence demandeur doivent donner leur accord.
La Commission Peer Review envoie ses conclusions argumentées (accord-refus-report) au Collège des médecins-directeurs.
Le Collège des médecins-directeurs prend la décision d’une intervention de l’assurance obligatoire, éventuellement sur base de l’avis remis par la Commission Peer Review et ce, pour chaque bénéficiaire individuellement.
Le Collège des médecins-directeurs communique sa décision motivée dans les trente jours ouvrables. Si ce Collège a demandé un avis de la Commission peer-review, il communique sa décision motivée dans les trente jours ouvrables après réception du rapport de la Commission Peer Review. Cette décision est communiquée à l’épileptologue du centre de référence concerné et qui a adhéré à la convention, au bénéficiaire concerné via son organisme assureur et au pharmacien hospitalier.
Les documents desquels il ressort qu'il est satisfait à l’une des indications mentionnées au point 3. doivent être conservés dans le dossier médical du bénéficiaire.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
Une copie de cette dérogation sera fournie au Collège des médecins-directeurs.
Dans ce cas, la prestation 171614-171625 doit être attestée.
6.2. Remplacement
Dans le cas d’un renouvellement d’un dispositif qui a déjà fait l’objet d’une intervention de l’assurance obligatoire dans le cadre de cette convention, la demande pour les prestations 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 et 171813-171824 peut être introduite par l’épileptologue après implantation au Collège des médecins-directeurs au moyen du formulaire de demande B-Form-I-08.
La demande contient notamment un rapport médical de l’évolution, dans lequel doit être mentionné entre autres le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171636-171640 doit être attestée.
6.3 Remplacement anticipé
La procédure expliquée au point 6.2. doit être appliquée.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171651-171662 doit être attestée.
6.4 Dérogation à la procédure
Pour les bénéficiaires qui ont déjà été implantés avant l’entrée en vigueur de la présente convention et qui remplissaient avant implantation toutes les conditions visées au point 3, une intervention de l’assurance obligatoire pour le renouvellement des neurostimulateurs et accessoires peut être accordée suivant les modalités prévues au point 6.1.
Dans ce cas particulier, l’équipe du centre de référence fait parvenir un dossier de demande d’intervention de l’assurance obligatoire pour un renouvellement au Collège des médecins-directeurs sur base des formulaires B-Form-I-07 et 08. Ce dossier comprendra les documents de la première implantation démontrant que cette implantation répondait aux critères d’intervention de l’assurance obligatoire ainsi qu’un rapport médical de l’évolution, dans lequel doit être entre autres mentionné le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Le Collège prendra la décision d’une intervention de l’assurance obligatoire pour le renouvellement, éventuellement sur base de l’avis de la Commission Peer Review.
7. Règles d’attestation
7.1 Règles de cumul et de non-cumul
Une intervention de l'assurance obligatoire pour la prestation 171496-171500, 171555-171566 ou 171614-171625 exclut, pendant une période de deux ans, une intervention de l'assurance pour la prestation 170892-170903.
7.2. Autres règles
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 suivent les modalités de remboursement de la catégorie A.
Les prestations 171614-171625, 171636-171640 ou 171651-171662 suivent les modalités de remboursement de la catégorie F.
L’intervention de l’assurance obligatoire pour la prestation 171511-171522 ne peut être accordée que minimum deux ans après la prestation 171496-171500, 171511-171522 ou 171533-171544.
L’intervention de l’assurance obligatoire pour la prestation 171570-171581 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 ou 171651-171662.
L’intervention de l’assurance obligatoire pour la prestation 171636-171640 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603,171614-171625, 171636-171640 ou 171651-171662..
7.3 Dérogation aux règles d’attestation
Pas d’application.
8. Engagements de l’établissement hospitalier
8.1. L’établissement hospitalier qui a adhéré à la convention est tenu de tenir à jour consciencieusement les caractéristiques de référence (baseline) et les données de suivi des bénéficiaires traités dans le cadre de cette convention afin de réaliser l'analyse prévue au point 9.2. Ces données doivent être conservées dans le Dossier Patient Informatisé.
8.2. Le pouvoir organisateur de l'établissement hospitalier porte la responsabilité de communiquer, sans délai, chaque modification au Fonctionnaire dirigeant du Service des soins de santé de l'INAMI et bien entendu à tous ceux qui sont concernés, dont en premier lieu les bénéficiaires.
Par e-mail au Secrétariat de la Commission de remboursement des implants et des dispositifs médicaux invasifs, à l’adresse suivante : implant@riziv-inami.fgov.be.
Lorsque le Fonctionnaire dirigeant constate des manquements, il informe les organismes assureurs du fait que le dispositif n’est plus remboursable pour cet établissement hospitalier.
9. Analyse
9.1. L’analyse des données émanant de cette application clinique limitée est effectuée par la Commission Peer Review et les organismes assureurs – nommés également évaluateur – qui ont signé la convention. Ils apportent un support technique et scientifique et évaluent les résultats du dispositif médical selon des critères précis.
9.2. Analyse – Rapport final
Au plus tard 6 mois avant la fin de la convention, l’évaluateur doit rédiger un rapport final sur base des données collectées et le transmettre à la Commission.
Ce rapport concerne tous les bénéficiaires implantés avec un neurostimulateur pour stimulation cérébrale profonde en cas d’épilepsie réfractaire depuis la date d’entrée en vigueur du remboursement.
Ce rapport final comprend au minimum les éléments suivants :
1) le nombre de patients traités, sexe, âge ;
2) analyse de tous les critères d’inclusion et d’exclusion dont indications, contre-indications, durée des symptômes, traitements, autres affections psychiatriques comorbides ;
3) résultats de la neurostimulation :
a. modification de la sévérité des crises
b. modification de la fréquence des crises
c. taux de répondeurs (= diminution de 50% ou plus de la fréquence des crises)
d. modification du traitement médicamenteux
4) effets secondaires, complications
5) une analyse rétrospective des coûts médicaux directs réalisée avec une comparaison des coûts cumulatifs pendant une période de un an avant l’implantation du stimulateur DBS avec les coûts cumulatifs de l’année suivant l’implantation. Les coûts étudiés seront les suivants :
a. nombre et durée des admissions en établissement hospitalier et liées à l’épilepsie. Les examens techniques relatifs au diagnostic et au traitement sont inclus, les coûts
dus à l’évaluation préchirurgicale ne font pas l’objet de l’étude
b. nombre de visite aux urgences
c. nombre de visite chez le médecin généraliste ou le neurologue
d. nombre et dose de spécialités pharmaceutiques antiépileptiques
6) une analyse des coûts indirects
7) une comparaison des résultats avec la littérature existante
Les organismes assureurs fourniront l’analyse rétrospective des coûts médicaux directs et indirects comme défini aux points 5 et 6 du contenu du rapport final.
Si ce rapport n’est pas communiqué à la date mentionnée ci-dessus, la Commission en informe le Ministre. Celui-ci peut prendre la décision de suspendre le remboursement du dispositif.
La Commission utilisera le rapport final qui évalue le dispositif comme base pour rédiger un règlement définitif. Ce règlement sera soumis au Ministre par l’intermédiaire de la Commission.
10. Droit de résiliation pour chaque partie prenante
La convention entre en vigueur le 1er décembre 2020 et est valable jusqu’au 30 novembre 2023 inclus mais peut toujours être résiliée par l’INAMI ou par un établissement hospitalier par lettre recommandée à la poste, adressée à l'autre partie, en respectant le délai de résiliation de trois mois qui prend cours le premier jour du mois suivant la date d'envoi de la lettre recommandée.
La convention expire dès que l’établissement hospitalier ne répond plus aux dispositions de cette convention.
11. Divers
A la demande de la Commission ou de l’évaluateur, une réunion peut être organisée à tout moment.
Teneinde een tijdelijke tegemoetkoming van de verplichte verzekering te kunnen genieten voor de verstrekkingen betreffende de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie moet aan volgende voorwaarden worden voldaan:
1. Doel van de overeenkomst
Deze overeenkomst heeft tot doel de tijdelijke tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging inzake de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie alsook de modaliteiten ervan te bepalen in het kader van een beperkte klinische toepassing gedurende de evaluatieperiode die loopt van 1 december 2020 tot en met 30 november 2023. Gedurende die periode wordt het hulpmiddel geëvalueerd volgens de bepalingen voorzien in punt 9.
2. Criteria betreffende de verplegingsinrichting
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien ze zijn uitgevoerd in een verplegingsinrichting die aan de volgende criteria voldoet en die de overeenkomst B-BKT-001-bis heeft afgesloten met het Verzekeringscomité:
De verplegingsinrichting moet gedurende de volledige looptijd van de overeenkomst voldoen aan de onderstaande criteria.
2.1. Enkel de verplegingsinrichtingen die de revalidatieovereenkomst van de referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden, hebben ondertekend, kunnen toetreden tot deze overeenkomst.
De verplegingsinrichting moet 24 uur op 24 en 7 dagen op 7 een neurochirurgische en neurologische permanentie hebben.
De indicatiestelling, de prechirurgische evaluatie, de implantatie, met inbegrip van de vervangingen, de revalidatie en een follow-up op lange termijn mogen enkel door deze verplegingsinrichtingen worden uitgevoerd. Het Comité van de verzekering voor geneeskundige verzorging stelt, op voorstel van de Dienst voor geneeskundige verzorging, een lijst op die continu wordt bijgewerkt, met vermelding van de samenstelling van het team per verplegingsinrichting en bezorgt die lijst ter informatie aan de Commissie.
De verplegingsinrichting en de artsen toegetreden tot de overeenkomst B-BKT-001-bis engageren zich tot het meewerken aan de evaluatie zoals bedoeld in punt 9.
De dagelijkse follow-up van de rechthebbende mag buiten die referentiecentra worden verricht.
2.2. Kandidatuurformulier voor de verplegingsinrichting
De verplegingsinrichting moet zich kenbaar maken bij de Dienst voor Geneeskundige verzorging op basis van het formulier B-Form-II-02.
2.3. Samenwerkingsakkoord
De verplegingsinrichting mag geen geformaliseerd samenwerkingsakkoord met andere verplegingsinrichtingen of met andere ziekenhuisverenigingen sluiten. Bovendien moet de ingreep binnen de muren van het bevoegde verplegingsinrichting worden uitgevoerd.
3. Criteria betreffende de rechthebbende
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien de rechthebbende aan de volgende criteria voldoet:
3.1. Inclusiecriteria
1) De rechthebbende lijdt aan focale epilepsie met focale complexe aanvallen met of zonder secundaire generalisering.
2) De rechthebbende lijdt aan refractaire epilepsie, d.w.z. waarbij de aanvallen onvoldoende onder controle kunnen worden gehouden met één van de potentieel doeltreffende anti-epileptica, die alleen of in combinatie worden toegediend, met optimale therapeutische dosissen waaraan geen onaanvaardbare bijwerkingen zijn verbonden; de aanvallen kunnen bovendien arbeidsongeschiktheid of een handicap tot gevolg hebben.
3) De rechthebbende moet een optimale farmacologische behandeling hebben gekregen. De rechthebbende is niet in staat om de epilepsie voldoende onder controle te houden met minstens 3 verschillende therapieën, waarvan minstens één combinatie, met optimale dosissen en gedurende een periode die volstaat om de doeltreffendheid ervan te beoordelen.
4) De rechthebbende komt niet in aanmerking voor chirurgie of de epilepsiechirurgie is een mislukking. De prechirurgische evaluatie omvat de volgende tests:
a. langdurige EEG-videomonitoring met registratie van de aanvallen
b. hogeresolutie-MRI van de hersenen
c. FDG-PET-scan van de hersenen
d. neuropsychologische evaluatie die de volgende elementen bevat:
i. IQ
ii. geheugenfuncties
iii.
e. psychiatrische evaluatie die onder andere de volgende elementen bevat:
i. Beck Depression Inventory
ii. QOLIE-31
In geval dit onderzoek niet gerealiseerd kan worden, bijvoorbeeld als gevolg van een mentale achterstand (die op zich geen tegenindicatie vormt voor de implantatie van een stimulator voor diepe hersenstimulatie), dient de reden duidelijk te worden vermeld op het formulier.
5) De rechthebbende moet op het moment van de implantatie minstens 18 jaar en maximaal 65 jaar oud zijn.
6) De rechthebbende kan een patiëntenprogrameerapparaat gebruiken en is bereid om zich aan de gevraagde tests te onderwerpen.
7) Alleen de rechthebbenden die duidelijk in staat zijn om, uit eigen wil, via een informed consent over de implantatie van elektroden en een neurostimulator te beslissen, komen in aanmerking. In die verklaring moeten omstandig de voor- en nadelen van de behandeling, de risico’s en de psychosociale impact worden toegelicht.
8) Als de rechthebbende met nervus vagusstimulatie wordt behandeld en indien daarmee onvoldoende resultaten worden bereikt, moet de rechthebbende vanaf de primo-implantatie minstens 2 jaar op de implantatie van een DBS-neurostimulator wachten en vice versa.
9) De gestimuleerde zone (doelgebied) is de zone die in de indicaties wordt vermeld en die door de huidige CE-markering wordt gedekt.
3.2. Exclusiecriteria
1) Ernstige neurologische of medische aandoening die een contra-indicatie is voor een cerebrale ingreep.
2) Elke chirurgische contra-indicatie om DBS te ondergaan, met inbegrip van de contra-indicaties die voor de DBS en/of voor de uitvoering van een preoperatieve MRI bekend zijn, contra-indicaties in het kader van een ingreep onder anesthesie of andere risicofactoraren voor een chirurgische ingreep (een ernstige cardiovasculaire aandoening, coagulopathie, …).
3) Ongepast gebruik van een product (alcohol, drugs, …)/middelenmisbruik waardoor het toestel niet correct kan worden gebruikt of waardoor er geen systematische medische/psychiatrische follow-up mogelijk is.
4) Suïcidale gedachten
5) Chronische psychotische problematiek die niet gestabiliseerd is onder behandeling, met uitzondering van een peri-ictale psychose
4. Criteria betreffende het hulpmiddel
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien het hulpmiddel aan de volgende criteria voldoet:
4.1. Definitie
De hulpmiddelen bedoeld onder de verstrekkingen 171614-171625, 171636-171640 en 171651-171662 dragen geen CE-markering, maar moeten het voorwerp uitmaken van een derogatie toegekend door de Minister die bevoegd is voor Volksgezondheid.
4.2. Criteria
Om te kunnen worden opgenomen op de nominatieve lijst voor de 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 en 171695-171706 moet het hulpmiddel beantwoorden aan de volgende criteria:
De werkzaamheid en veiligheid van het hulpmiddel moeten zijn aangetoond met behulp van klinische studies. Deze studies dienen gepubliceerd te zijn in een internationaal erkend peer reviewed tijdschrift. Onder de gerealiseerde klinische studies, moet er minstens een follow-up studie zijn met minimum 100 patiënten die gedurende minimaal 2 jaar gevolgd worden, evenals een vergelijkende, gerandomiseerde klinische studie gedurende minimum 3 maand en met een a priori vastgelegde statistische power van minstens 80%. De follow-up studie en de gerandomiseerde studie mogen dezelfde studie zijn.
4.3. Garantievoorwaarden
Om te kunnen worden opgenomen op de nominatieve lijst voor de verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 en 171813-171824 moet het hulpmiddel beantwoorden aan de volgende garantievoorwaarden:
- Niet heroplaadbare neurostimulatoren:
een totale garantie van 24 maanden moet worden gegeven voor de niet heroplaadbare neurostimulatoren
- Heroplaadbare neurostimulatoren:
een garantie van negen jaar moet worden gegeven voor de heroplaadbare neurostimulatoren: een volledige garantie voor de eerste vijf jaar en voor de volgende vier jaar een garantie pro rata. Voor de lader 171791-171802 en 171813-171824 is een volledige garantie van negen jaar vereist. De verstrekkingen 171614-171625, 171636-171640 en 171651-171662, moeten ook aan de garantievoorwaarden voldoen.
5. Aantal rechthebbenden
Het aantal rechthebbenden dat voor een tegemoetkoming van de verplichte verzekering in aanmerking kan komen, wordt beperkt tot 25 nieuwe rechthebbenden per jaar.
Zodra het aantal dreigt te worden overschreden, brengt het Secretariaat de Commissie op de hoogte en wordt aan de evaluator gevraagd een verslag te bezorgen. De aard van het verslag wordt vastgelegd door de Commissie.
De Commissie brengt de verplegingsinrichtingen en de verstrekkers van het betrokken hulpmiddel op de hoogte en neemt de noodzakelijke maatregelen.
6. Aanvraagprocedure en formulieren
6.1. Eerste implantatie
De aanvraag tot tegemoetkoming van de verplichte verzekering voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 gebeurt als volgt:
De terugbetalingsaanvraag voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 wordt vóór de implantatie op basis van formulier B-Form-I-07 ingediend door de epileptoloog aan het College van artsen-directeurs.
Die verstrekkingen mogen enkel na akkoord van het College van artsen-directeurs door de verplichte verzekering worden terugbetaald. In geval van twijfel legt het College van artsen-directeurs de aanvraag voor advies voor aan de Commissie Peer Review, die bestaat uit zorgverleners die de revalidatieovereenkomst “referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden” hebben ondertekend. Die Commissie Peer Review moet de aanvraag van het team van het referentiecentrum binnen de drie maanden onderzoeken. De Commissie Peer Review informeert het team van het referentiecentrum dat de aanvraag heeft ingediend, zodat deze de aanvraag kan verdedigen. Tijdens de bespreking van de dossiers die in aanmerking komen voor een tegemoetkoming van de verplichte verzekering voor neurostimulatie kan er altijd een lid van het College van artsen-directeurs of een arts, lid van de Commissie, aanwezig zijn. Minstens twee leden van de Commissie Peer Review die tot twee andere verplegingsinrichtingen dan het aanvragend referentiecentrum behoren, moeten hun akkoord geven.
De Commissie Peer Review bezorgt haar geargumenteerde conclusies (akkoord – weigering – uitstel) aan het College van artsen-directeurs.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging, eventueel op basis van het advies van de Commissie Peer Review, en dit voor elke rechthebbende afzonderlijk.
Het College van artsen-directeurs deelt zijn gemotiveerde beslissing mee binnen de dertig werkdagen. Als dit College een advies van de Commissie Peer Review heeft gevraagd, deelt het zijn gemotiveerde beslissing mee binnen de dertig werkdagen na ontvangst van het verslag van de Commissie Peer Review. Die beslissing wordt aan de epileptoloog van het betrokken referentiecentrum dat tot de overeenkomst is toegetreden, aan de betrokken rechthebbende via zijn verzekeringsinstelling en aan de ziekenhuisapotheker meegedeeld.
De documenten, waaruit blijkt dat voldaan is aan één van de indicaties vermeld onder punt 3, moeten steeds in het medisch dossier van de rechthebbende aanwezig zijn.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171614-171625 worden geattesteerd.
6.2. Vervanging
In geval van hernieuwing van een hulpmiddel dat reeds het voorwerp heeft uitgemaakt van een tegemoetkoming van de verplichte verzekering in het kader van deze overeenkomst, mag de aanvraag voor de verstrekkingen 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 en 171813-171824 na implantatie ingediend worden door de epileptoloog bij het College van artsen-directeurs door middel van het aanvraagformulier B-Form-I-08.
De aanvraag bevat onder andere een medisch evolutieverslag, waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de redenen voor de noodzaak van de hernieuwing zijn opgenomen.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171636-171640 worden geattesteerd.
6.3. Voortijdige vervanging
De procedure beschreven onder punt 6.2. dient te worden toegepast.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171651-171662 worden geattesteerd.
6.4. Derogatie aan de procedure
Voor de rechthebbenden die reeds vóór de inwerkingtreding van deze overeenkomst ingeplant zijn en die vóór de implantatie aan alle voorwaarden bedoeld in punt 3 voldeden, kan een tegemoetkoming van de verplichte verzekering voor de hernieuwing van de neurostimulatoren en toebehoren worden toegekend volgens de modaliteiten voorzien in punt 6.1.
In dat geval bezorgt het team van het referentiecentrum een aanvraagdossier tot tegemoetkoming van de verplichte verzekering voor een hernieuwing aan het College van artsen-directeurs op basis van de formulieren B-Form-I-07 en 08. Dat dossier bevat de documenten van de eerste implantatie waarin wordt aangetoond dat deze implantatie aan de vergoedingscriteria van de verplichte verzekering voldeed en een medisch evolutieverslag waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de rechtvaardiging van de hernieuwing zijn opgenomen.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor de hernieuwing, eventueel op basis van het advies van de Commissie Peer Review.
7. Regels voor attestering
7.1. Cumul en non-cumulregels
Een tegemoetkoming van de verplichte verzekering voor de verstrekking 171496-171500, 171555-171566 of 171614-171625 sluit tijdens een periode van twee jaar een tegemoetkoming van de verzekering voor de verstrekking 170892-170903 uit.
7.2. Andere regels
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 volgen de vergoedingsmodaliteiten van categorie A.
De verstrekkingen 171614-171625, 171636-171640 en 171651-171662 volgen de vergoedingsmodaliteiten van categorie F.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171511-171522 kan pas minstens twee jaar na de verstrekking 171496-171500, 171511-171522 of 171533-171544 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171570-171581 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 of 171651-171662 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171695-171706 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171625, 171636-171640 of 171651-171662 worden toegekend.
7.3. Derogatie van de attesteringsregels
Niet van toepassing.
8. Verbintenissen van de verplegingsinrichting
8.1. De verplegingsinrichting die tot de overeenkomst is toegetreden moet de baseline karakteristieken en de follow-up gegevens van de rechthebbenden die in het kader van deze overeenkomst zijn behandeld nauwgezet bijhouden om de analyse voorzien in punt 9.2. uit te voeren. Deze gegevens moeten in het Elektronisch Patiënten Dossier bewaard worden.
8.2. De inrichtende macht van de verplegingsinrichting is verantwoordelijk voor de onverwijlde mededeling van elke wijziging aan de leidend ambtenaar van de Dienst voor geneeskundige verzorging van het RIZIV en natuurlijk aan alle betrokkenen, waaronder in de eerste plaats de rechthebbenden.
Per e-mail aan het Secretariaat van de Commissie Tegemoetkoming Implantaten en Invasieve Medische Hulpmiddelen, op het volgende adres: implant@riziv-inami.fgov.be.
Wanneer de Leidend ambtenaar nalatigheden vaststelt, brengt hij de verzekeringsinstellingen ervan op de hoogte dat het hulpmiddel voor die verplegingsinrichting niet meer mag worden terugbetaald.
9. Analyse
9.1. De analyse van de gegevens uit deze beperkte klinische toepassing wordt uitgevoerd door de Commissie Peer Review en de verzekeringsinstellingen – verder evaluator genoemd – die mee de overeenkomst ondertekend hebben. Zij verlenen technische en wetenschappelijke ondersteuning en analyseren de resultaten van het medisch hulpmiddel volgens precieze criteria.
9.2. Analyse – Eindverslag
Uiterlijk 6 maanden voor het einde van de overeenkomst moet de evaluator op basis van de verzamelde gegevens een eindverslag opstellen en aan de Commissie bezorgen.
Dit verslag betreft alle rechthebbenden geïmplanteerd met een neurostimulator voor diepe hersenstimulatie in geval van refractaire epilepsie sinds de datum van inwerkingtreding van de terugbetaling.
Dat eindverslag moet minstens de volgende elementen bevatten:
1) het aantal behandelde patiënten, geslacht, leeftijd;
2) analyse van alle inclusie- en exclusiecriteria, waaronder indicaties, contra-indicaties, duur van de symptomen, behandelingen, andere comorbide psychiatrische aandoeningen;
3) resultaten van de neurostimulatie:
a. wijziging van de ernst van de aanvallen
b. wijziging van de aanvalsfrequentie
c. responsratio (= daling met 50 % of meer van de aanvalsfrequentie)
d. wijziging van de medicamenteuze behandeling
4) bijwerkingen, complicaties;
5) een retrospectieve analyse van de directe medische kosten, gerealiseerd met een vergelijking van de cumulatieve kosten gedurende één jaar vóór de implantatie van een DBS-stimulator met de cumulatieve kosten van het jaar na implantatie. De volgende kosten worden bestudeerd:
a. aantal en duur van opnames in verplegingsinrichting en gelinkt aan epilepsie. De technische onderzoeken met betrekking tot diagnostiek of behandeling zijn inbegrepen.
De kosten te wijten aan de prechirurgische evaluatie zijn niet het onderwerp van de studie.
b. aantal bezoeken aan de spoed
c. aantal bezoeken bij de huisarts of de neuroloog
d. aantal en dosis van de anti-epileptische farmaceutische specialiteiten
6) een analyse van de indirecte kosten;
7) een vergelijking van de resultaten met de bestaande literatuur.
De verzekeringsinstellingen zullen de retrospectieve analyse van de directe en indirecte medische kosten zoals gedefinieerd in punten 5 en 6 van de inhoud van het eindverslag bezorgen.
Indien dit verslag niet op de voormelde datum wordt meegedeeld, brengt de Commissie de minister daarvan op de hoogte. Deze kan beslissen om de terugbetaling van het hulpmiddel stop te zetten.
De Commissie zal het eindverslag waarin het hulpmiddel wordt geëvalueerd, als basis kunnen gebruiken voor het opstellen van een definitieve regeling. Die regeling zal door de Commissie aan de Minister worden voorgelegd.
10. Opzeggingsrecht voor elke betrokken partij
De overeenkomst treedt in werking op 1 december 2020 en is geldig tot en met 30 november 2023 maar kan steeds door het RIZIV of door een verplegingsinrichting worden opgezegd met een ter post aangetekende brief die aan de andere partij wordt gericht, mits inachtneming van een opzeggingstermijn van drie maanden die ingaat op de eerste dag van de maand volgend op de datum van verzending van de aangetekende brief.
De overeenkomst verstrijkt zodra de verplegingsinrichting niet meer aan de bepalingen van deze overeenkomst voldoet.
11. Varia
Op verzoek van de Commissie of van de evaluator kan er op elk moment een vergadering worden georganiseerd.
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Afin de pouvoir bénéficier d’une intervention temporaire de l’assurance obligatoire pour les prestations relatives aux neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire, il doit être satisfait aux conditions suivantes :
1. But de la convention
Cette convention a pour but de fixer le remboursement temporaire de l’assurance obligatoire soins de santé ainsi que ses modalités concernant les neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire dans le cadre d’une application clinique limitée pendant la période d’évaluation qui cours du 1er décembre 2020 au 30 novembre 2023 inclus. Pendant cette période, le dispositif sera évalué selon les dispositions prévues au point 9.
2. Critères concernant l’établissement hospitalier
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si elles sont effectuées dans un établissement hospitalier qui répond aux critères suivants et qui a conclu la convention B-ACL-001-bis avec le Comité de l’assurance:
L’établissement hospitalier doit durant la totalité de la durée de la convention répondre aux critères ci-dessous.
2.1. Seuls les établissements hospitaliers ayant signé la convention de rééducation des centres de référence pour bénéficiaires souffrant d’épilepsie rebelle peuvent adhérer à cette convention.
L’établissement hospitalier doit avoir une permanence en neurochirurgie et en neurologie 24 heures sur 24 et 7 jours sur 7.
La pose d’indication, l’évaluation pré-chirurgicale, l’implantation, y compris les remplacements, la rééducation et un suivi à long terme ne peuvent être effectuées que par ces seuls établissements hospitaliers. Le Comité de l'assurance soins de santé dresse, sur proposition du Service des soins de santé, une liste, mise à jour de façon continue, avec la composition de l'équipe par établissement hospitalier et l'envoie pour information à la Commission.
L’établissement hospitalier et les médecins ayant adhéré à la convention B-ACL-001-bis s’engagent à collaborer à l’évaluation visée au point 9.
Le suivi du bénéficiaire au quotidien est permis hors de ces centres de référence.
2.2. Formulaire de candidature pour l’établissement hospitalier
L’établissement hospitalier doit se faire connaître auprès du Service des Soins de santé sur base du formulaire B-Form-II-02
2.3. Accord de coopération
L’établissement hospitalier ne peut conclure ni d’accord de coopération formalisé avec d’autres établissements hospitaliers ni d’associations hospitalières. De plus, l’intervention doit être réalisée dans les murs de l’établissement hospitalier habilité.
3. Critères concernant le bénéficiaire
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le bénéficiaire répond aux critères suivants :
3.1. Critères d’inclusion
1) Le bénéficiaire est atteint d’épilepsie focale avec des crises focales complexes, avec ou sans généralisation secondaire.
2) Le bénéficiaire est atteint d´épilepsie réfractaire, c´est-à-dire qu’un contrôle satisfaisant des crises ne peut pas être obtenu avec un des médicaments antiépileptiques potentiellement efficaces, administré seul ou en combinaison, à des doses thérapeutiques optimales et non associées à des effets secondaires inacceptables, les crises entrainant des incapacités et un handicap.
3) Le bénéficiaire doit avoir été traité par un traitement pharmacologique optimal. Le bénéficiaire n’a pas de contrôle satisfaisant de l’épilepsie avec au minimum 3 thérapies différentes, dont au minimum une association, aux doses optimales et durant une période suffisante pour en apprécier l’efficacité.
4) Le bénéficiaire n’est pas éligible pour une chirurgie ou la chirurgie de l’épilepsie est un échec. L’évaluation préchirurgicale inclut les tests suivants :
a. Enregistrement vidéo-EEG de longue durée avec enregistrement des crises
b. IRM à haute résolution du cerveau
c. FDG-PET du cerveau
d. Évaluation neuropsychologique incluant les éléments suivants :
i. QI
ii. mémoire
iii. fonctions exécutives frontales
e. Évaluation psychiatrique incluant entre autres les éléments suivants :
i. inventaire de dépression de Beck
ii. QoLIE-31
Au cas où un examen ne serait pas réalisable, par exemple suite à un retard mental (qui ne constitue pas en soi une contre-indication à l´implantation d´un stimulateur pour stimulation cérébrale profonde), la raison doit en être clairement mentionnée dans le formulaire.
5) Le bénéficiaire doit être âgé d’au moins 18 ans et de maximum 65 ans lors de l’implantation.
6) Le bénéficiaire est capable d’utiliser un programmateur et de se soumettre aux tests demandés
7) Seuls les bénéficiaires clairement capables, de décider de leur plein gré, via une déclaration de consentement éclairé, de l’implantation d’électrodes et d’un neurostimulateur entrent en ligne de compte. Cette déclaration doit expliquer de manière détaillée les avantages et inconvénients du traitement, les risques ainsi que l’impact psychosocial.
8) Si le bénéficiaire est traité par stimulation du nerf vague et en cas de résultats insuffisants, il faut minimum 2 ans depuis la primo-implantation pour pouvoir implanter un neurostimulateur DBS et inversement.
9) La zone stimulée (cible) est celle qui est reprise dans les indications couvertes par le marquage CE en vigueur.
3.2. Critères d’exclusion
1) Affection neurologique ou médicale grave qui constitue une contre-indication pour une intervention cérébrale.
2) Toute contre-indication chirurgicale pour subir une DBS, y compris les contre-indications connues pour la DBS et/ou pour l’exécution d’une IRM préopératoire, contre-indications dans le cadre d’une intervention sous anesthésie ou autres facteurs à risque pour une intervention chirurgicale (une affection cardio-vasculaire grave, coagulopathie,…).
3) Utilisation inappropriée d’un produit (alcool, drogue,…), abus de substance qui ne permet pas un usage correct de l’appareil ou rendant un suivi médical/psychiatrique systématique impossible.
4) Idées suicidaires
5) Problématique chronique psychotique non stabilisée sous traitement, à l’exception de la psychose péri-ictale
4. Critères concernant le dispositif
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le dispositif répond aux critères suivants:
4.1. Définition
Les dispositifs visés par les prestations 171614-171625, 171636-171640 et 171651-171662 ne portent pas le marquage CE mais doivent faire l'objet d'une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
4.2. Critères
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 et 171695-171706, le dispositif doit répondre aux critères suivants :
Le dispositif doit avoir fait preuve de son efficacité et sa sécurité à l’aide d’ études cliniques. Ces études seront publiées dans une revue « peer-reviewed » reconnue internationalement. Parmi les études cliniques réalisées, il faut au minimum une étude de suivi sur au minimum 100 patients suivis pendant minimum 2 ans, ainsi qu’une étude clinique comparative, randomisée au minimum pendant 3 mois et ayant une puissance statistique déterminée a priori d’au moins 80%. L’étude de suivi et l’étude randomisée peuvent être la même étude.
Un dispositif qui est une adaptation d’un dispositif déjà inscrit sur la liste nominative pour le même distributeur, sans changement du mode d’action et sans impact négatif sur l’efficacité, la sécurité et la qualité, peut être inscrit sans études cliniques à condition que le distributeur décrive les adaptations et leurs conséquences pratiques.
4.3. Conditions de garantie
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 et 171813-171824, le dispositif doit répondre aux conditions de garantie suivantes :
- neurostimulateurs non-rechargeables :
une garantie totale de 24 mois doit être donnée pour les neurostimulateurs non-rechargeables
- neurostimulateurs rechargeables:
une garantie de neuf ans doit être donnée pour les neurostimulateurs rechargeables : une garantie totale pour les cinq premières années et pour les quatre années suivantes une garantie au prorata. Pour le chargeur 171791-171802 et 171813-171824, une garantie totale de neuf ans est exigée. Les prestations 171614-171625, 171636-171640 et 171651-171662 doivent également répondre à ces conditions de garantie.
5. Nombre de bénéficiaires
Le nombre de bénéficiaires qui peuvent entrer en ligne de compte pour une intervention de l’assurance obligatoire est limité à 25 nouveaux bénéficiaires par an.
Dès que le nombre menace d’être dépassé, le Secrétariat en informe la Commission et il est demandé à l’évaluateur de communiquer un rapport. La nature du rapport est établie par la Commission.
La Commission informe les établissements hospitaliers et les distributeurs du dispositif concerné et prend les mesures nécessaires.
6. Procédure de demande et formulaires
6.1 Première implantation
La demande d’intervention de l’assurance obligatoire pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 se déroule comme suit:
La demande d’intervention pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 est introduite avant implantation par l’épileptologue sur base du formulaire B-Form-I-07 au Collège des médecins-directeurs.
Ces prestations ne peuvent faire l’objet d’une intervention de l’assurance qu’après accord du Collège des médecins-directeurs. En cas de doute, le Collège des médecins-directeurs peut transmettre la demande pour avis à la Commission Peer Review, composée des prestataires ayant signé la convention de rééducation « centres de référence pour bénéficiaires souffrant d’épilepsie réfractaire ». Cette Commission Peer Review est alors tenue d’examiner la demande de l’équipe du centre de référence dans les trois mois. La Commission Peer Review avertit l’équipe du centre de référence qui a introduit la demande afin de lui permettre de la défendre. Au cours de la discussion portant sur les dossiers qui entrent en ligne de compte pour une intervention de l’assurance pour la neurostimulation, un membre du Collège des médecins-directeurs ou un médecin, membre de la Commission, peut toujours être présent. Minimum deux membres de la Commission Peer Review issus de deux établissements autres que le centre de référence demandeur doivent donner leur accord.
La Commission Peer Review envoie ses conclusions argumentées (accord-refus-report) au Collège des médecins-directeurs.
Le Collège des médecins-directeurs prend la décision d’une intervention de l’assurance obligatoire, éventuellement sur base de l’avis remis par la Commission Peer Review et ce, pour chaque bénéficiaire individuellement.
Le Collège des médecins-directeurs communique sa décision motivée dans les trente jours ouvrables. Si ce Collège a demandé un avis de la Commission peer-review, il communique sa décision motivée dans les trente jours ouvrables après réception du rapport de la Commission Peer Review. Cette décision est communiquée à l’épileptologue du centre de référence concerné et qui a adhéré à la convention, au bénéficiaire concerné via son organisme assureur et au pharmacien hospitalier.
Les documents desquels il ressort qu'il est satisfait à l’une des indications mentionnées au point 3. doivent être conservés dans le dossier médical du bénéficiaire.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
Une copie de cette dérogation sera fournie au Collège des médecins-directeurs.
Dans ce cas, la prestation 171614-171625 doit être attestée.
6.2. Remplacement
Dans le cas d’un renouvellement d’un dispositif qui a déjà fait l’objet d’une intervention de l’assurance obligatoire dans le cadre de cette convention, la demande pour les prestations 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 et 171813-171824 peut être introduite par l’épileptologue après implantation au Collège des médecins-directeurs au moyen du formulaire de demande B-Form-I-08.
La demande contient notamment un rapport médical de l’évolution, dans lequel doit être mentionné entre autres le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171636-171640 doit être attestée.
6.3 Remplacement anticipé
La procédure expliquée au point 6.2. doit être appliquée.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171651-171662 doit être attestée.
6.4 Dérogation à la procédure
Pour les bénéficiaires qui ont déjà été implantés avant l’entrée en vigueur de la présente convention et qui remplissaient avant implantation toutes les conditions visées au point 3, une intervention de l’assurance obligatoire pour le renouvellement des neurostimulateurs et accessoires peut être accordée suivant les modalités prévues au point 6.1.
Dans ce cas particulier, l’équipe du centre de référence fait parvenir un dossier de demande d’intervention de l’assurance obligatoire pour un renouvellement au Collège des médecins-directeurs sur base des formulaires B-Form-I-07 et 08. Ce dossier comprendra les documents de la première implantation démontrant que cette implantation répondait aux critères d’intervention de l’assurance obligatoire ainsi qu’un rapport médical de l’évolution, dans lequel doit être entre autres mentionné le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Le Collège prendra la décision d’une intervention de l’assurance obligatoire pour le renouvellement, éventuellement sur base de l’avis de la Commission Peer Review.
7. Règles d’attestation
7.1 Règles de cumul et de non-cumul
Une intervention de l'assurance obligatoire pour la prestation 171496-171500, 171555-171566 ou 171614-171625 exclut, pendant une période de deux ans, une intervention de l'assurance pour la prestation 170892-170903.
7.2. Autres règles
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 suivent les modalités de remboursement de la catégorie A.
Les prestations 171614-171625, 171636-171640 ou 171651-171662 suivent les modalités de remboursement de la catégorie F.
L’intervention de l’assurance obligatoire pour la prestation 171511-171522 ne peut être accordée que minimum deux ans après la prestation 171496-171500, 171511-171522 ou 171533-171544.
L’intervention de l’assurance obligatoire pour la prestation 171570-171581 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 ou 171651-171662.
L’intervention de l’assurance obligatoire pour la prestation 171636-171640 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603,171614-171625, 171636-171640 ou 171651-171662..
7.3 Dérogation aux règles d’attestation
Pas d’application.
8. Engagements de l’établissement hospitalier
8.1. L’établissement hospitalier qui a adhéré à la convention est tenu de tenir à jour consciencieusement les caractéristiques de référence (baseline) et les données de suivi des bénéficiaires traités dans le cadre de cette convention afin de réaliser l'analyse prévue au point 9.2. Ces données doivent être conservées dans le Dossier Patient Informatisé.
8.2. Le pouvoir organisateur de l'établissement hospitalier porte la responsabilité de communiquer, sans délai, chaque modification au Fonctionnaire dirigeant du Service des soins de santé de l'INAMI et bien entendu à tous ceux qui sont concernés, dont en premier lieu les bénéficiaires.
Par e-mail au Secrétariat de la Commission de remboursement des implants et des dispositifs médicaux invasifs, à l’adresse suivante : implant@riziv-inami.fgov.be.
Lorsque le Fonctionnaire dirigeant constate des manquements, il informe les organismes assureurs du fait que le dispositif n’est plus remboursable pour cet établissement hospitalier.
9. Analyse
9.1. L’analyse des données émanant de cette application clinique limitée est effectuée par la Commission Peer Review et les organismes assureurs – nommés également évaluateur – qui ont signé la convention. Ils apportent un support technique et scientifique et évaluent les résultats du dispositif médical selon des critères précis.
9.2. Analyse – Rapport final
Au plus tard 6 mois avant la fin de la convention, l’évaluateur doit rédiger un rapport final sur base des données collectées et le transmettre à la Commission.
Ce rapport concerne tous les bénéficiaires implantés avec un neurostimulateur pour stimulation cérébrale profonde en cas d’épilepsie réfractaire depuis la date d’entrée en vigueur du remboursement.
Ce rapport final comprend au minimum les éléments suivants :
1) le nombre de patients traités, sexe, âge ;
2) analyse de tous les critères d’inclusion et d’exclusion dont indications, contre-indications, durée des symptômes, traitements, autres affections psychiatriques comorbides ;
3) résultats de la neurostimulation :
a. modification de la sévérité des crises
b. modification de la fréquence des crises
c. taux de répondeurs (= diminution de 50% ou plus de la fréquence des crises)
d. modification du traitement médicamenteux
4) effets secondaires, complications
5) une analyse rétrospective des coûts médicaux directs réalisée avec une comparaison des coûts cumulatifs pendant une période de un an avant l’implantation du stimulateur DBS avec les coûts cumulatifs de l’année suivant l’implantation. Les coûts étudiés seront les suivants :
a. nombre et durée des admissions en établissement hospitalier et liées à l’épilepsie. Les examens techniques relatifs au diagnostic et au traitement sont inclus, les coûts
dus à l’évaluation préchirurgicale ne font pas l’objet de l’étude
b. nombre de visite aux urgences
c. nombre de visite chez le médecin généraliste ou le neurologue
d. nombre et dose de spécialités pharmaceutiques antiépileptiques
6) une analyse des coûts indirects
7) une comparaison des résultats avec la littérature existante
Les organismes assureurs fourniront l’analyse rétrospective des coûts médicaux directs et indirects comme défini aux points 5 et 6 du contenu du rapport final.
Si ce rapport n’est pas communiqué à la date mentionnée ci-dessus, la Commission en informe le Ministre. Celui-ci peut prendre la décision de suspendre le remboursement du dispositif.
La Commission utilisera le rapport final qui évalue le dispositif comme base pour rédiger un règlement définitif. Ce règlement sera soumis au Ministre par l’intermédiaire de la Commission.
10. Droit de résiliation pour chaque partie prenante
La convention entre en vigueur le 1er décembre 2020 et est valable jusqu’au 30 novembre 2023 inclus mais peut toujours être résiliée par l’INAMI ou par un établissement hospitalier par lettre recommandée à la poste, adressée à l'autre partie, en respectant le délai de résiliation de trois mois qui prend cours le premier jour du mois suivant la date d'envoi de la lettre recommandée.
La convention expire dès que l’établissement hospitalier ne répond plus aux dispositions de cette convention.
11. Divers
A la demande de la Commission ou de l’évaluateur, une réunion peut être organisée à tout moment.
Teneinde een tijdelijke tegemoetkoming van de verplichte verzekering te kunnen genieten voor de verstrekkingen betreffende de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie moet aan volgende voorwaarden worden voldaan:
1. Doel van de overeenkomst
Deze overeenkomst heeft tot doel de tijdelijke tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging inzake de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie alsook de modaliteiten ervan te bepalen in het kader van een beperkte klinische toepassing gedurende de evaluatieperiode die loopt van 1 december 2020 tot en met 30 november 2023. Gedurende die periode wordt het hulpmiddel geëvalueerd volgens de bepalingen voorzien in punt 9.
2. Criteria betreffende de verplegingsinrichting
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien ze zijn uitgevoerd in een verplegingsinrichting die aan de volgende criteria voldoet en die de overeenkomst B-BKT-001-bis heeft afgesloten met het Verzekeringscomité:
De verplegingsinrichting moet gedurende de volledige looptijd van de overeenkomst voldoen aan de onderstaande criteria.
2.1. Enkel de verplegingsinrichtingen die de revalidatieovereenkomst van de referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden, hebben ondertekend, kunnen toetreden tot deze overeenkomst.
De verplegingsinrichting moet 24 uur op 24 en 7 dagen op 7 een neurochirurgische en neurologische permanentie hebben.
De indicatiestelling, de prechirurgische evaluatie, de implantatie, met inbegrip van de vervangingen, de revalidatie en een follow-up op lange termijn mogen enkel door deze verplegingsinrichtingen worden uitgevoerd. Het Comité van de verzekering voor geneeskundige verzorging stelt, op voorstel van de Dienst voor geneeskundige verzorging, een lijst op die continu wordt bijgewerkt, met vermelding van de samenstelling van het team per verplegingsinrichting en bezorgt die lijst ter informatie aan de Commissie.
De verplegingsinrichting en de artsen toegetreden tot de overeenkomst B-BKT-001-bis engageren zich tot het meewerken aan de evaluatie zoals bedoeld in punt 9.
De dagelijkse follow-up van de rechthebbende mag buiten die referentiecentra worden verricht.
2.2. Kandidatuurformulier voor de verplegingsinrichting
De verplegingsinrichting moet zich kenbaar maken bij de Dienst voor Geneeskundige verzorging op basis van het formulier B-Form-II-02.
2.3. Samenwerkingsakkoord
De verplegingsinrichting mag geen geformaliseerd samenwerkingsakkoord met andere verplegingsinrichtingen of met andere ziekenhuisverenigingen sluiten. Bovendien moet de ingreep binnen de muren van het bevoegde verplegingsinrichting worden uitgevoerd.
3. Criteria betreffende de rechthebbende
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien de rechthebbende aan de volgende criteria voldoet:
3.1. Inclusiecriteria
1) De rechthebbende lijdt aan focale epilepsie met focale complexe aanvallen met of zonder secundaire generalisering.
2) De rechthebbende lijdt aan refractaire epilepsie, d.w.z. waarbij de aanvallen onvoldoende onder controle kunnen worden gehouden met één van de potentieel doeltreffende anti-epileptica, die alleen of in combinatie worden toegediend, met optimale therapeutische dosissen waaraan geen onaanvaardbare bijwerkingen zijn verbonden; de aanvallen kunnen bovendien arbeidsongeschiktheid of een handicap tot gevolg hebben.
3) De rechthebbende moet een optimale farmacologische behandeling hebben gekregen. De rechthebbende is niet in staat om de epilepsie voldoende onder controle te houden met minstens 3 verschillende therapieën, waarvan minstens één combinatie, met optimale dosissen en gedurende een periode die volstaat om de doeltreffendheid ervan te beoordelen.
4) De rechthebbende komt niet in aanmerking voor chirurgie of de epilepsiechirurgie is een mislukking. De prechirurgische evaluatie omvat de volgende tests:
a. langdurige EEG-videomonitoring met registratie van de aanvallen
b. hogeresolutie-MRI van de hersenen
c. FDG-PET-scan van de hersenen
d. neuropsychologische evaluatie die de volgende elementen bevat:
i. IQ
ii. geheugenfuncties
iii.
e. psychiatrische evaluatie die onder andere de volgende elementen bevat:
i. Beck Depression Inventory
ii. QOLIE-31
In geval dit onderzoek niet gerealiseerd kan worden, bijvoorbeeld als gevolg van een mentale achterstand (die op zich geen tegenindicatie vormt voor de implantatie van een stimulator voor diepe hersenstimulatie), dient de reden duidelijk te worden vermeld op het formulier.
5) De rechthebbende moet op het moment van de implantatie minstens 18 jaar en maximaal 65 jaar oud zijn.
6) De rechthebbende kan een patiëntenprogrameerapparaat gebruiken en is bereid om zich aan de gevraagde tests te onderwerpen.
7) Alleen de rechthebbenden die duidelijk in staat zijn om, uit eigen wil, via een informed consent over de implantatie van elektroden en een neurostimulator te beslissen, komen in aanmerking. In die verklaring moeten omstandig de voor- en nadelen van de behandeling, de risico’s en de psychosociale impact worden toegelicht.
8) Als de rechthebbende met nervus vagusstimulatie wordt behandeld en indien daarmee onvoldoende resultaten worden bereikt, moet de rechthebbende vanaf de primo-implantatie minstens 2 jaar op de implantatie van een DBS-neurostimulator wachten en vice versa.
9) De gestimuleerde zone (doelgebied) is de zone die in de indicaties wordt vermeld en die door de huidige CE-markering wordt gedekt.
3.2. Exclusiecriteria
1) Ernstige neurologische of medische aandoening die een contra-indicatie is voor een cerebrale ingreep.
2) Elke chirurgische contra-indicatie om DBS te ondergaan, met inbegrip van de contra-indicaties die voor de DBS en/of voor de uitvoering van een preoperatieve MRI bekend zijn, contra-indicaties in het kader van een ingreep onder anesthesie of andere risicofactoraren voor een chirurgische ingreep (een ernstige cardiovasculaire aandoening, coagulopathie, …).
3) Ongepast gebruik van een product (alcohol, drugs, …)/middelenmisbruik waardoor het toestel niet correct kan worden gebruikt of waardoor er geen systematische medische/psychiatrische follow-up mogelijk is.
4) Suïcidale gedachten
5) Chronische psychotische problematiek die niet gestabiliseerd is onder behandeling, met uitzondering van een peri-ictale psychose
4. Criteria betreffende het hulpmiddel
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien het hulpmiddel aan de volgende criteria voldoet:
4.1. Definitie
De hulpmiddelen bedoeld onder de verstrekkingen 171614-171625, 171636-171640 en 171651-171662 dragen geen CE-markering, maar moeten het voorwerp uitmaken van een derogatie toegekend door de Minister die bevoegd is voor Volksgezondheid.
4.2. Criteria
Om te kunnen worden opgenomen op de nominatieve lijst voor de 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 en 171695-171706 moet het hulpmiddel beantwoorden aan de volgende criteria:
De werkzaamheid en veiligheid van het hulpmiddel moeten zijn aangetoond met behulp van klinische studies. Deze studies dienen gepubliceerd te zijn in een internationaal erkend peer reviewed tijdschrift. Onder de gerealiseerde klinische studies, moet er minstens een follow-up studie zijn met minimum 100 patiënten die gedurende minimaal 2 jaar gevolgd worden, evenals een vergelijkende, gerandomiseerde klinische studie gedurende minimum 3 maand en met een a priori vastgelegde statistische power van minstens 80%. De follow-up studie en de gerandomiseerde studie mogen dezelfde studie zijn.
Een hulpmiddel dat een aanpassing is van een hulpmiddel dat reeds op de nominatieve lijst voor dezelfde verdeler ingeschreven is, zonder wijziging van het werkingsmechanisme en zonder negatieve impact op de werkzaamheid, de veiligheid en de kwaliteit, kan ingeschreven worden zonder klinische studies op voorwaarde dat de verdeler de aanpassingen en hun praktische consequenties beschrijft.
4.3. Garantievoorwaarden
Om te kunnen worden opgenomen op de nominatieve lijst voor de verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 en 171813-171824 moet het hulpmiddel beantwoorden aan de volgende garantievoorwaarden:
- Niet heroplaadbare neurostimulatoren:
een totale garantie van 24 maanden moet worden gegeven voor de niet heroplaadbare neurostimulatoren
- Heroplaadbare neurostimulatoren:
een garantie van negen jaar moet worden gegeven voor de heroplaadbare neurostimulatoren: een volledige garantie voor de eerste vijf jaar en voor de volgende vier jaar een garantie pro rata. Voor de lader 171791-171802 en 171813-171824 is een volledige garantie van negen jaar vereist. De verstrekkingen 171614-171625, 171636-171640 en 171651-171662, moeten ook aan de garantievoorwaarden voldoen.
5. Aantal rechthebbenden
Het aantal rechthebbenden dat voor een tegemoetkoming van de verplichte verzekering in aanmerking kan komen, wordt beperkt tot 25 nieuwe rechthebbenden per jaar.
Zodra het aantal dreigt te worden overschreden, brengt het Secretariaat de Commissie op de hoogte en wordt aan de evaluator gevraagd een verslag te bezorgen. De aard van het verslag wordt vastgelegd door de Commissie.
De Commissie brengt de verplegingsinrichtingen en de verstrekkers van het betrokken hulpmiddel op de hoogte en neemt de noodzakelijke maatregelen.
6. Aanvraagprocedure en formulieren
6.1. Eerste implantatie
De aanvraag tot tegemoetkoming van de verplichte verzekering voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 gebeurt als volgt:
De terugbetalingsaanvraag voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 wordt vóór de implantatie op basis van formulier B-Form-I-07 ingediend door de epileptoloog aan het College van artsen-directeurs.
Die verstrekkingen mogen enkel na akkoord van het College van artsen-directeurs door de verplichte verzekering worden terugbetaald. In geval van twijfel legt het College van artsen-directeurs de aanvraag voor advies voor aan de Commissie Peer Review, die bestaat uit zorgverleners die de revalidatieovereenkomst “referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden” hebben ondertekend. Die Commissie Peer Review moet de aanvraag van het team van het referentiecentrum binnen de drie maanden onderzoeken. De Commissie Peer Review informeert het team van het referentiecentrum dat de aanvraag heeft ingediend, zodat deze de aanvraag kan verdedigen. Tijdens de bespreking van de dossiers die in aanmerking komen voor een tegemoetkoming van de verplichte verzekering voor neurostimulatie kan er altijd een lid van het College van artsen-directeurs of een arts, lid van de Commissie, aanwezig zijn. Minstens twee leden van de Commissie Peer Review die tot twee andere verplegingsinrichtingen dan het aanvragend referentiecentrum behoren, moeten hun akkoord geven.
De Commissie Peer Review bezorgt haar geargumenteerde conclusies (akkoord – weigering – uitstel) aan het College van artsen-directeurs.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging, eventueel op basis van het advies van de Commissie Peer Review, en dit voor elke rechthebbende afzonderlijk.
Het College van artsen-directeurs deelt zijn gemotiveerde beslissing mee binnen de dertig werkdagen. Als dit College een advies van de Commissie Peer Review heeft gevraagd, deelt het zijn gemotiveerde beslissing mee binnen de dertig werkdagen na ontvangst van het verslag van de Commissie Peer Review. Die beslissing wordt aan de epileptoloog van het betrokken referentiecentrum dat tot de overeenkomst is toegetreden, aan de betrokken rechthebbende via zijn verzekeringsinstelling en aan de ziekenhuisapotheker meegedeeld.
De documenten, waaruit blijkt dat voldaan is aan één van de indicaties vermeld onder punt 3, moeten steeds in het medisch dossier van de rechthebbende aanwezig zijn.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171614-171625 worden geattesteerd.
6.2. Vervanging
In geval van hernieuwing van een hulpmiddel dat reeds het voorwerp heeft uitgemaakt van een tegemoetkoming van de verplichte verzekering in het kader van deze overeenkomst, mag de aanvraag voor de verstrekkingen 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 en 171813-171824 na implantatie ingediend worden door de epileptoloog bij het College van artsen-directeurs door middel van het aanvraagformulier B-Form-I-08.
De aanvraag bevat onder andere een medisch evolutieverslag, waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de redenen voor de noodzaak van de hernieuwing zijn opgenomen.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171636-171640 worden geattesteerd.
6.3. Voortijdige vervanging
De procedure beschreven onder punt 6.2. dient te worden toegepast.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171651-171662 worden geattesteerd.
6.4. Derogatie aan de procedure
Voor de rechthebbenden die reeds vóór de inwerkingtreding van deze overeenkomst ingeplant zijn en die vóór de implantatie aan alle voorwaarden bedoeld in punt 3 voldeden, kan een tegemoetkoming van de verplichte verzekering voor de hernieuwing van de neurostimulatoren en toebehoren worden toegekend volgens de modaliteiten voorzien in punt 6.1.
In dat geval bezorgt het team van het referentiecentrum een aanvraagdossier tot tegemoetkoming van de verplichte verzekering voor een hernieuwing aan het College van artsen-directeurs op basis van de formulieren B-Form-I-07 en 08. Dat dossier bevat de documenten van de eerste implantatie waarin wordt aangetoond dat deze implantatie aan de vergoedingscriteria van de verplichte verzekering voldeed en een medisch evolutieverslag waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de rechtvaardiging van de hernieuwing zijn opgenomen.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor de hernieuwing, eventueel op basis van het advies van de Commissie Peer Review.
7. Regels voor attestering
7.1. Cumul en non-cumulregels
Een tegemoetkoming van de verplichte verzekering voor de verstrekking 171496-171500, 171555-171566 of 171614-171625 sluit tijdens een periode van twee jaar een tegemoetkoming van de verzekering voor de verstrekking 170892-170903 uit.
7.2. Andere regels
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 volgen de vergoedingsmodaliteiten van categorie A.
De verstrekkingen 171614-171625, 171636-171640 en 171651-171662 volgen de vergoedingsmodaliteiten van categorie F.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171511-171522 kan pas minstens twee jaar na de verstrekking 171496-171500, 171511-171522 of 171533-171544 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171570-171581 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 of 171651-171662 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171695-171706 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171625, 171636-171640 of 171651-171662 worden toegekend.
7.3. Derogatie van de attesteringsregels
Niet van toepassing.
8. Verbintenissen van de verplegingsinrichting
8.1. De verplegingsinrichting die tot de overeenkomst is toegetreden moet de baseline karakteristieken en de follow-up gegevens van de rechthebbenden die in het kader van deze overeenkomst zijn behandeld nauwgezet bijhouden om de analyse voorzien in punt 9.2. uit te voeren. Deze gegevens moeten in het Elektronisch Patiënten Dossier bewaard worden.
8.2. De inrichtende macht van de verplegingsinrichting is verantwoordelijk voor de onverwijlde mededeling van elke wijziging aan de leidend ambtenaar van de Dienst voor geneeskundige verzorging van het RIZIV en natuurlijk aan alle betrokkenen, waaronder in de eerste plaats de rechthebbenden.
Per e-mail aan het Secretariaat van de Commissie Tegemoetkoming Implantaten en Invasieve Medische Hulpmiddelen, op het volgende adres: implant@riziv-inami.fgov.be.
Wanneer de Leidend ambtenaar nalatigheden vaststelt, brengt hij de verzekeringsinstellingen ervan op de hoogte dat het hulpmiddel voor die verplegingsinrichting niet meer mag worden terugbetaald.
9. Analyse
9.1. De analyse van de gegevens uit deze beperkte klinische toepassing wordt uitgevoerd door de Commissie Peer Review en de verzekeringsinstellingen – verder evaluator genoemd – die mee de overeenkomst ondertekend hebben. Zij verlenen technische en wetenschappelijke ondersteuning en analyseren de resultaten van het medisch hulpmiddel volgens precieze criteria.
9.2. Analyse – Eindverslag
Uiterlijk 6 maanden voor het einde van de overeenkomst moet de evaluator op basis van de verzamelde gegevens een eindverslag opstellen en aan de Commissie bezorgen.
Dit verslag betreft alle rechthebbenden geïmplanteerd met een neurostimulator voor diepe hersenstimulatie in geval van refractaire epilepsie sinds de datum van inwerkingtreding van de terugbetaling.
Dat eindverslag moet minstens de volgende elementen bevatten:
1) het aantal behandelde patiënten, geslacht, leeftijd;
2) analyse van alle inclusie- en exclusiecriteria, waaronder indicaties, contra-indicaties, duur van de symptomen, behandelingen, andere comorbide psychiatrische aandoeningen;
3) resultaten van de neurostimulatie:
a. wijziging van de ernst van de aanvallen
b. wijziging van de aanvalsfrequentie
c. responsratio (= daling met 50 % of meer van de aanvalsfrequentie)
d. wijziging van de medicamenteuze behandeling
4) bijwerkingen, complicaties;
5) een retrospectieve analyse van de directe medische kosten, gerealiseerd met een vergelijking van de cumulatieve kosten gedurende één jaar vóór de implantatie van een DBS-stimulator met de cumulatieve kosten van het jaar na implantatie. De volgende kosten worden bestudeerd:
a. aantal en duur van opnames in verplegingsinrichting en gelinkt aan epilepsie. De technische onderzoeken met betrekking tot diagnostiek of behandeling zijn inbegrepen.
De kosten te wijten aan de prechirurgische evaluatie zijn niet het onderwerp van de studie.
b. aantal bezoeken aan de spoed
c. aantal bezoeken bij de huisarts of de neuroloog
d. aantal en dosis van de anti-epileptische farmaceutische specialiteiten
6) een analyse van de indirecte kosten;
7) een vergelijking van de resultaten met de bestaande literatuur.
De verzekeringsinstellingen zullen de retrospectieve analyse van de directe en indirecte medische kosten zoals gedefinieerd in punten 5 en 6 van de inhoud van het eindverslag bezorgen.
Indien dit verslag niet op de voormelde datum wordt meegedeeld, brengt de Commissie de minister daarvan op de hoogte. Deze kan beslissen om de terugbetaling van het hulpmiddel stop te zetten.
De Commissie zal het eindverslag waarin het hulpmiddel wordt geëvalueerd, als basis kunnen gebruiken voor het opstellen van een definitieve regeling. Die regeling zal door de Commissie aan de Minister worden voorgelegd.
10. Opzeggingsrecht voor elke betrokken partij
De overeenkomst treedt in werking op 1 december 2020 en is geldig tot en met 30 november 2023 maar kan steeds door het RIZIV of door een verplegingsinrichting worden opgezegd met een ter post aangetekende brief die aan de andere partij wordt gericht, mits inachtneming van een opzeggingstermijn van drie maanden die ingaat op de eerste dag van de maand volgend op de datum van verzending van de aangetekende brief.
De overeenkomst verstrijkt zodra de verplegingsinrichting niet meer aan de bepalingen van deze overeenkomst voldoet.
11. Varia
Op verzoek van de Commissie of van de evaluator kan er op elk moment een vergadering worden georganiseerd.
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Afin de pouvoir bénéficier d’une intervention temporaire de l’assurance obligatoire pour les prestations relatives aux neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire, il doit être satisfait aux conditions suivantes :
1. But de la convention
Cette convention a pour but de fixer le remboursement temporaire de l’assurance obligatoire soins de santé ainsi que ses modalités concernant les neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire dans le cadre d’une application clinique limitée pendant la période d’évaluation qui cours du 1er décembre 2020 au 30 novembre 2023 inclus. Pendant cette période, le dispositif sera évalué selon les dispositions prévues au point 9.
2. Critères concernant l’établissement hospitalier
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si elles sont effectuées dans un établissement hospitalier qui répond aux critères suivants et qui a conclu la convention B-ACL-001-bis avec le Comité de l’assurance:
L’établissement hospitalier doit durant la totalité de la durée de la convention répondre aux critères ci-dessous.
2.1. Seuls les établissements hospitaliers ayant signé la convention de rééducation des centres de référence pour bénéficiaires souffrant d’épilepsie rebelle peuvent adhérer à cette convention.
L’établissement hospitalier doit avoir une permanence en neurochirurgie et en neurologie 24 heures sur 24 et 7 jours sur 7.
La pose d’indication, l’évaluation pré-chirurgicale, l’implantation, y compris les remplacements, la rééducation et un suivi à long terme ne peuvent être effectuées que par ces seuls établissements hospitaliers. Le Comité de l'assurance soins de santé dresse, sur proposition du Service des soins de santé, une liste, mise à jour de façon continue, avec la composition de l'équipe par établissement hospitalier et l'envoie pour information à la Commission.
L’établissement hospitalier et les médecins ayant adhéré à la convention B-ACL-001-bis s’engagent à collaborer à l’évaluation visée au point 9.
Le suivi du bénéficiaire au quotidien est permis hors de ces centres de référence.
2.2. Formulaire de candidature pour l’établissement hospitalier
L’établissement hospitalier doit se faire connaître auprès du Service des Soins de santé sur base du formulaire B-Form-II-02
2.3. Accord de coopération
L’établissement hospitalier ne peut conclure ni d’accord de coopération formalisé avec d’autres établissements hospitaliers ni d’associations hospitalières. De plus, l’intervention doit être réalisée dans les murs de l’établissement hospitalier habilité.
3. Critères concernant le bénéficiaire
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le bénéficiaire répond aux critères suivants :
3.1. Critères d’inclusion
1) Le bénéficiaire est atteint d’épilepsie focale avec des crises focales complexes, avec ou sans généralisation secondaire.
2) Le bénéficiaire est atteint d´épilepsie réfractaire, c´est-à-dire qu’un contrôle satisfaisant des crises ne peut pas être obtenu avec un des médicaments antiépileptiques potentiellement efficaces, administré seul ou en combinaison, à des doses thérapeutiques optimales et non associées à des effets secondaires inacceptables, les crises entrainant des incapacités et un handicap.
3) Le bénéficiaire doit avoir été traité par un traitement pharmacologique optimal. Le bénéficiaire n’a pas de contrôle satisfaisant de l’épilepsie avec au minimum 3 thérapies différentes, dont au minimum une association, aux doses optimales et durant une période suffisante pour en apprécier l’efficacité.
4) Le bénéficiaire n’est pas éligible pour une chirurgie ou la chirurgie de l’épilepsie est un échec. L’évaluation préchirurgicale inclut les tests suivants :
a. Enregistrement vidéo-EEG de longue durée avec enregistrement des crises
b. IRM à haute résolution du cerveau
c. FDG-PET du cerveau
d. Évaluation neuropsychologique incluant les éléments suivants :
i. QI
ii. mémoire
iii. fonctions exécutives frontales
e. Évaluation psychiatrique incluant entre autres les éléments suivants :
i. inventaire de dépression de Beck
ii. QoLIE-31
Au cas où un examen ne serait pas réalisable, par exemple suite à un retard mental (qui ne constitue pas en soi une contre-indication à l´implantation d´un stimulateur pour stimulation cérébrale profonde), la raison doit en être clairement mentionnée dans le formulaire.
5) Le bénéficiaire doit être âgé d’au moins 18 ans et de maximum 65 ans lors de l’implantation.
6) Le bénéficiaire est capable d’utiliser un programmateur et de se soumettre aux tests demandés
7) Seuls les bénéficiaires clairement capables, de décider de leur plein gré, via une déclaration de consentement éclairé, de l’implantation d’électrodes et d’un neurostimulateur entrent en ligne de compte. Cette déclaration doit expliquer de manière détaillée les avantages et inconvénients du traitement, les risques ainsi que l’impact psychosocial.
8) Si le bénéficiaire est traité par stimulation du nerf vague et en cas de résultats insuffisants, il faut minimum 2 ans depuis la primo-implantation pour pouvoir implanter un neurostimulateur DBS et inversement.
9) La zone stimulée (cible) est celle qui est reprise dans les indications couvertes par le marquage CE en vigueur.
3.2. Critères d’exclusion
1) Affection neurologique ou médicale grave qui constitue une contre-indication pour une intervention cérébrale.
2) Toute contre-indication chirurgicale pour subir une DBS, y compris les contre-indications connues pour la DBS et/ou pour l’exécution d’une IRM préopératoire, contre-indications dans le cadre d’une intervention sous anesthésie ou autres facteurs à risque pour une intervention chirurgicale (une affection cardio-vasculaire grave, coagulopathie,…).
3) Utilisation inappropriée d’un produit (alcool, drogue,…), abus de substance qui ne permet pas un usage correct de l’appareil ou rendant un suivi médical/psychiatrique systématique impossible.
4) Idées suicidaires
5) Problématique chronique psychotique non stabilisée sous traitement, à l’exception de la psychose péri-ictale
4. Critères concernant le dispositif
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le dispositif répond aux critères suivants:
4.1. Définition
Les dispositifs visés par les prestations 171614-171625, 171636-171640 et 171651-171662 ne portent pas le marquage CE mais doivent faire l'objet d'une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
4.2. Critères
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 et 171695-171706, le dispositif doit répondre aux critères suivants :
Le dispositif doit avoir fait preuve de son efficacité et sa sécurité à l’aide d’ études cliniques. Ces études seront publiées dans une revue « peer-reviewed » reconnue internationalement. Parmi les études cliniques réalisées, il faut au minimum une étude de suivi sur au minimum 100 patients suivis pendant minimum 2 ans, ainsi qu’une étude clinique comparative, randomisée au minimum pendant 3 mois et ayant une puissance statistique déterminée a priori d’au moins 80%. L’étude de suivi et l’étude randomisée peuvent être la même étude.
Un dispositif qui est une adaptation d’un dispositif déjà inscrit sur la liste nominative pour le même distributeur, sans changement du mode d’action et sans impact négatif sur l’efficacité, la sécurité et la qualité, peut être inscrit sans études cliniques à condition que le distributeur décrive les adaptations et leurs conséquences pratiques.
4.3. Conditions de garantie
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 et 171813-171824, le dispositif doit répondre aux conditions de garantie suivantes :
- neurostimulateurs non-rechargeables :
une garantie totale de 24 mois doit être donnée pour les neurostimulateurs non-rechargeables
- neurostimulateurs rechargeables:
une garantie de neuf ans doit être donnée pour les neurostimulateurs rechargeables : une garantie totale pour les cinq premières années et pour les quatre années suivantes une garantie au prorata. Pour le chargeur 171791-171802 et 171813-171824, une garantie totale de neuf ans est exigée. Les prestations 171614-171625, 171636-171640 et 171651-171662 doivent également répondre à ces conditions de garantie.
5. Nombre de bénéficiaires
Le nombre de bénéficiaires qui peuvent entrer en ligne de compte pour une intervention de l’assurance obligatoire est limité à 25 nouveaux bénéficiaires par an.
Dès que le nombre menace d’être dépassé, le Secrétariat en informe la Commission et il est demandé à l’évaluateur de communiquer un rapport. La nature du rapport est établie par la Commission.
La Commission informe les établissements hospitaliers et les distributeurs du dispositif concerné et prend les mesures nécessaires.
6. Procédure de demande et formulaires
6.1 Première implantation
La demande d’intervention de l’assurance obligatoire pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 se déroule comme suit:
La demande d’intervention pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 est introduite avant implantation par l’épileptologue sur base du formulaire B-Form-I-07 au Collège des médecins-directeurs.
Ces prestations ne peuvent faire l’objet d’une intervention de l’assurance qu’après accord du Collège des médecins-directeurs. En cas de doute, le Collège des médecins-directeurs peut transmettre la demande pour avis à la Commission Peer Review, composée des prestataires ayant signé la convention de rééducation « centres de référence pour bénéficiaires souffrant d’épilepsie réfractaire ». Cette Commission Peer Review est alors tenue d’examiner la demande de l’équipe du centre de référence dans les trois mois. La Commission Peer Review avertit l’équipe du centre de référence qui a introduit la demande afin de lui permettre de la défendre. Au cours de la discussion portant sur les dossiers qui entrent en ligne de compte pour une intervention de l’assurance pour la neurostimulation, un membre du Collège des médecins-directeurs ou un médecin, membre de la Commission, peut toujours être présent. Minimum deux membres de la Commission Peer Review issus de deux établissements autres que le centre de référence demandeur doivent donner leur accord.
La Commission Peer Review envoie ses conclusions argumentées (accord-refus-report) au Collège des médecins-directeurs.
Le Collège des médecins-directeurs prend la décision d’une intervention de l’assurance obligatoire, éventuellement sur base de l’avis remis par la Commission Peer Review et ce, pour chaque bénéficiaire individuellement.
Le Collège des médecins-directeurs communique sa décision motivée dans les trente jours ouvrables. Si ce Collège a demandé un avis de la Commission peer-review, il communique sa décision motivée dans les trente jours ouvrables après réception du rapport de la Commission Peer Review. Cette décision est communiquée à l’épileptologue du centre de référence concerné et qui a adhéré à la convention, au bénéficiaire concerné via son organisme assureur et au pharmacien hospitalier.
Les documents desquels il ressort qu'il est satisfait à l’une des indications mentionnées au point 3. doivent être conservés dans le dossier médical du bénéficiaire.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
Une copie de cette dérogation sera fournie au Collège des médecins-directeurs.
Dans ce cas, la prestation 171614-171625 doit être attestée.
6.2. Remplacement
Dans le cas d’un renouvellement d’un dispositif qui a déjà fait l’objet d’une intervention de l’assurance obligatoire dans le cadre de cette convention, la demande pour les prestations 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 et 171813-171824 peut être introduite par l’épileptologue après implantation au Collège des médecins-directeurs au moyen du formulaire de demande B-Form-I-08.
La demande contient notamment un rapport médical de l’évolution, dans lequel doit être mentionné entre autres le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171636-171640 doit être attestée.
6.3 Remplacement anticipé
La procédure expliquée au point 6.2. doit être appliquée.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171651-171662 doit être attestée.
6.4 Dérogation à la procédure
Pour les bénéficiaires qui ont déjà été implantés avant l’entrée en vigueur de la présente convention et qui remplissaient avant implantation toutes les conditions visées au point 3, une intervention de l’assurance obligatoire pour le renouvellement des neurostimulateurs et accessoires peut être accordée suivant les modalités prévues au point 6.1.
Dans ce cas particulier, l’équipe du centre de référence fait parvenir un dossier de demande d’intervention de l’assurance obligatoire pour un renouvellement au Collège des médecins-directeurs sur base des formulaires B-Form-I-07 et 08. Ce dossier comprendra les documents de la première implantation démontrant que cette implantation répondait aux critères d’intervention de l’assurance obligatoire ainsi qu’un rapport médical de l’évolution, dans lequel doit être entre autres mentionné le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Le Collège prendra la décision d’une intervention de l’assurance obligatoire pour le renouvellement, éventuellement sur base de l’avis de la Commission Peer Review.
7. Règles d’attestation
7.1 Règles de cumul et de non-cumul
Une intervention de l'assurance obligatoire pour la prestation 171496-171500, 171555-171566 ou 171614-171625 exclut, pendant une période de deux ans, une intervention de l'assurance pour la prestation 170892-170903.
7.2. Autres règles
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 suivent les modalités de remboursement de la catégorie A.
Les prestations 171614-171625, 171636-171640 ou 171651-171662 suivent les modalités de remboursement de la catégorie F.
L’intervention de l’assurance obligatoire pour la prestation 171511-171522 ne peut être accordée que minimum deux ans après la prestation 171496-171500, 171511-171522 ou 171533-171544.
L’intervention de l’assurance obligatoire pour la prestation 171570-171581 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 ou 171651-171662.
L’intervention de l’assurance obligatoire pour la prestation 171636-171640 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603,171614-171625, 171636-171640 ou 171651-171662..
7.3 Dérogation aux règles d’attestation
Pas d’application.
8. Engagements de l’établissement hospitalier
8.1. L’établissement hospitalier qui a adhéré à la convention est tenu de tenir à jour consciencieusement les caractéristiques de référence (baseline) et les données de suivi des bénéficiaires traités dans le cadre de cette convention afin de réaliser l'analyse prévue au point 9.2. Ces données doivent être conservées dans le Dossier Patient Informatisé.
8.2. Le pouvoir organisateur de l'établissement hospitalier porte la responsabilité de communiquer, sans délai, chaque modification au Fonctionnaire dirigeant du Service des soins de santé de l'INAMI et bien entendu à tous ceux qui sont concernés, dont en premier lieu les bénéficiaires.
Par e-mail au Secrétariat de la Commission de remboursement des implants et des dispositifs médicaux invasifs, à l’adresse suivante : implant@riziv-inami.fgov.be.
Lorsque le Fonctionnaire dirigeant constate des manquements, il informe les organismes assureurs du fait que le dispositif n’est plus remboursable pour cet établissement hospitalier.
9. Analyse
9.1. L’analyse des données émanant de cette application clinique limitée est effectuée par la Commission Peer Review et les organismes assureurs – nommés également évaluateur – qui ont signé la convention. Ils apportent un support technique et scientifique et évaluent les résultats du dispositif médical selon des critères précis.
9.2. Analyse – Rapport final
Au plus tard 6 mois avant la fin de la convention, l’évaluateur doit rédiger un rapport final sur base des données collectées et le transmettre à la Commission.
Ce rapport concerne tous les bénéficiaires implantés avec un neurostimulateur pour stimulation cérébrale profonde en cas d’épilepsie réfractaire depuis la date d’entrée en vigueur du remboursement.
Ce rapport final comprend au minimum les éléments suivants :
1) le nombre de patients traités, sexe, âge ;
2) analyse de tous les critères d’inclusion et d’exclusion dont indications, contre-indications, durée des symptômes, traitements, autres affections psychiatriques comorbides ;
3) résultats de la neurostimulation :
a. modification de la sévérité des crises
b. modification de la fréquence des crises
c. taux de répondeurs (= diminution de 50% ou plus de la fréquence des crises)
d. modification du traitement médicamenteux
4) effets secondaires, complications
5) une analyse rétrospective des coûts médicaux directs réalisée avec une comparaison des coûts cumulatifs pendant une période de un an avant l’implantation du stimulateur DBS avec les coûts cumulatifs de l’année suivant l’implantation. Les coûts étudiés seront les suivants :
a. nombre et durée des admissions en établissement hospitalier et liées à l’épilepsie. Les examens techniques relatifs au diagnostic et au traitement sont inclus, les coûts
dus à l’évaluation préchirurgicale ne font pas l’objet de l’étude
b. nombre de visite aux urgences
c. nombre de visite chez le médecin généraliste ou le neurologue
d. nombre et dose de spécialités pharmaceutiques antiépileptiques
6) une analyse des coûts indirects
7) une comparaison des résultats avec la littérature existante
Les organismes assureurs fourniront l’analyse rétrospective des coûts médicaux directs et indirects comme défini aux points 5 et 6 du contenu du rapport final.
Si ce rapport n’est pas communiqué à la date mentionnée ci-dessus, la Commission en informe le Ministre. Celui-ci peut prendre la décision de suspendre le remboursement du dispositif.
La Commission utilisera le rapport final qui évalue le dispositif comme base pour rédiger un règlement définitif. Ce règlement sera soumis au Ministre par l’intermédiaire de la Commission.
10. Droit de résiliation pour chaque partie prenante
La convention entre en vigueur le 1er décembre 2020 et est valable jusqu’au 30 novembre 2023 inclus mais peut toujours être résiliée par l’INAMI ou par un établissement hospitalier par lettre recommandée à la poste, adressée à l'autre partie, en respectant le délai de résiliation de trois mois qui prend cours le premier jour du mois suivant la date d'envoi de la lettre recommandée.
La convention expire dès que l’établissement hospitalier ne répond plus aux dispositions de cette convention.
11. Divers
A la demande de la Commission ou de l’évaluateur, une réunion peut être organisée à tout moment.
Teneinde een tijdelijke tegemoetkoming van de verplichte verzekering te kunnen genieten voor de verstrekkingen betreffende de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie moet aan volgende voorwaarden worden voldaan:
1. Doel van de overeenkomst
Deze overeenkomst heeft tot doel de tijdelijke tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging inzake de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie alsook de modaliteiten ervan te bepalen in het kader van een beperkte klinische toepassing gedurende de evaluatieperiode die loopt van 1 december 2020 tot en met 30 november 2023. Gedurende die periode wordt het hulpmiddel geëvalueerd volgens de bepalingen voorzien in punt 9.
2. Criteria betreffende de verplegingsinrichting
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien ze zijn uitgevoerd in een verplegingsinrichting die aan de volgende criteria voldoet en die de overeenkomst B-BKT-001-bis heeft afgesloten met het Verzekeringscomité:
De verplegingsinrichting moet gedurende de volledige looptijd van de overeenkomst voldoen aan de onderstaande criteria.
2.1. Enkel de verplegingsinrichtingen die de revalidatieovereenkomst van de referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden, hebben ondertekend, kunnen toetreden tot deze overeenkomst.
De verplegingsinrichting moet 24 uur op 24 en 7 dagen op 7 een neurochirurgische en neurologische permanentie hebben.
De indicatiestelling, de prechirurgische evaluatie, de implantatie, met inbegrip van de vervangingen, de revalidatie en een follow-up op lange termijn mogen enkel door deze verplegingsinrichtingen worden uitgevoerd. Het Comité van de verzekering voor geneeskundige verzorging stelt, op voorstel van de Dienst voor geneeskundige verzorging, een lijst op die continu wordt bijgewerkt, met vermelding van de samenstelling van het team per verplegingsinrichting en bezorgt die lijst ter informatie aan de Commissie.
De verplegingsinrichting en de artsen toegetreden tot de overeenkomst B-BKT-001-bis engageren zich tot het meewerken aan de evaluatie zoals bedoeld in punt 9.
De dagelijkse follow-up van de rechthebbende mag buiten die referentiecentra worden verricht.
2.2. Kandidatuurformulier voor de verplegingsinrichting
De verplegingsinrichting moet zich kenbaar maken bij de Dienst voor Geneeskundige verzorging op basis van het formulier B-Form-II-02.
2.3. Samenwerkingsakkoord
De verplegingsinrichting mag geen geformaliseerd samenwerkingsakkoord met andere verplegingsinrichtingen of met andere ziekenhuisverenigingen sluiten. Bovendien moet de ingreep binnen de muren van het bevoegde verplegingsinrichting worden uitgevoerd.
3. Criteria betreffende de rechthebbende
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien de rechthebbende aan de volgende criteria voldoet:
3.1. Inclusiecriteria
1) De rechthebbende lijdt aan focale epilepsie met focale complexe aanvallen met of zonder secundaire generalisering.
2) De rechthebbende lijdt aan refractaire epilepsie, d.w.z. waarbij de aanvallen onvoldoende onder controle kunnen worden gehouden met één van de potentieel doeltreffende anti-epileptica, die alleen of in combinatie worden toegediend, met optimale therapeutische dosissen waaraan geen onaanvaardbare bijwerkingen zijn verbonden; de aanvallen kunnen bovendien arbeidsongeschiktheid of een handicap tot gevolg hebben.
3) De rechthebbende moet een optimale farmacologische behandeling hebben gekregen. De rechthebbende is niet in staat om de epilepsie voldoende onder controle te houden met minstens 3 verschillende therapieën, waarvan minstens één combinatie, met optimale dosissen en gedurende een periode die volstaat om de doeltreffendheid ervan te beoordelen.
4) De rechthebbende komt niet in aanmerking voor chirurgie of de epilepsiechirurgie is een mislukking. De prechirurgische evaluatie omvat de volgende tests:
a. langdurige EEG-videomonitoring met registratie van de aanvallen
b. hogeresolutie-MRI van de hersenen
c. FDG-PET-scan van de hersenen
d. neuropsychologische evaluatie die de volgende elementen bevat:
i. IQ
ii. geheugenfuncties
iii.
e. psychiatrische evaluatie die onder andere de volgende elementen bevat:
i. Beck Depression Inventory
ii. QOLIE-31
In geval dit onderzoek niet gerealiseerd kan worden, bijvoorbeeld als gevolg van een mentale achterstand (die op zich geen tegenindicatie vormt voor de implantatie van een stimulator voor diepe hersenstimulatie), dient de reden duidelijk te worden vermeld op het formulier.
5) De rechthebbende moet op het moment van de implantatie minstens 18 jaar en maximaal 65 jaar oud zijn.
6) De rechthebbende kan een patiëntenprogrameerapparaat gebruiken en is bereid om zich aan de gevraagde tests te onderwerpen.
7) Alleen de rechthebbenden die duidelijk in staat zijn om, uit eigen wil, via een informed consent over de implantatie van elektroden en een neurostimulator te beslissen, komen in aanmerking. In die verklaring moeten omstandig de voor- en nadelen van de behandeling, de risico’s en de psychosociale impact worden toegelicht.
8) Als de rechthebbende met nervus vagusstimulatie wordt behandeld en indien daarmee onvoldoende resultaten worden bereikt, moet de rechthebbende vanaf de primo-implantatie minstens 2 jaar op de implantatie van een DBS-neurostimulator wachten en vice versa.
9) De gestimuleerde zone (doelgebied) is de zone die in de indicaties wordt vermeld en die door de huidige CE-markering wordt gedekt.
3.2. Exclusiecriteria
1) Ernstige neurologische of medische aandoening die een contra-indicatie is voor een cerebrale ingreep.
2) Elke chirurgische contra-indicatie om DBS te ondergaan, met inbegrip van de contra-indicaties die voor de DBS en/of voor de uitvoering van een preoperatieve MRI bekend zijn, contra-indicaties in het kader van een ingreep onder anesthesie of andere risicofactoraren voor een chirurgische ingreep (een ernstige cardiovasculaire aandoening, coagulopathie, …).
3) Ongepast gebruik van een product (alcohol, drugs, …)/middelenmisbruik waardoor het toestel niet correct kan worden gebruikt of waardoor er geen systematische medische/psychiatrische follow-up mogelijk is.
4) Suïcidale gedachten
5) Chronische psychotische problematiek die niet gestabiliseerd is onder behandeling, met uitzondering van een peri-ictale psychose
4. Criteria betreffende het hulpmiddel
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien het hulpmiddel aan de volgende criteria voldoet:
4.1. Definitie
De hulpmiddelen bedoeld onder de verstrekkingen 171614-171625, 171636-171640 en 171651-171662 dragen geen CE-markering, maar moeten het voorwerp uitmaken van een derogatie toegekend door de Minister die bevoegd is voor Volksgezondheid.
4.2. Criteria
Om te kunnen worden opgenomen op de nominatieve lijst voor de 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 en 171695-171706 moet het hulpmiddel beantwoorden aan de volgende criteria:
De werkzaamheid en veiligheid van het hulpmiddel moeten zijn aangetoond met behulp van klinische studies. Deze studies dienen gepubliceerd te zijn in een internationaal erkend peer reviewed tijdschrift. Onder de gerealiseerde klinische studies, moet er minstens een follow-up studie zijn met minimum 100 patiënten die gedurende minimaal 2 jaar gevolgd worden, evenals een vergelijkende, gerandomiseerde klinische studie gedurende minimum 3 maand en met een a priori vastgelegde statistische power van minstens 80%. De follow-up studie en de gerandomiseerde studie mogen dezelfde studie zijn.
Een hulpmiddel dat een aanpassing is van een hulpmiddel dat reeds op de nominatieve lijst voor dezelfde verdeler ingeschreven is, zonder wijziging van het werkingsmechanisme en zonder negatieve impact op de werkzaamheid, de veiligheid en de kwaliteit, kan ingeschreven worden zonder klinische studies op voorwaarde dat de verdeler de aanpassingen en hun praktische consequenties beschrijft.
4.3. Garantievoorwaarden
Om te kunnen worden opgenomen op de nominatieve lijst voor de verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 en 171813-171824 moet het hulpmiddel beantwoorden aan de volgende garantievoorwaarden:
- Niet heroplaadbare neurostimulatoren:
een totale garantie van 24 maanden moet worden gegeven voor de niet heroplaadbare neurostimulatoren
- Heroplaadbare neurostimulatoren:
een garantie van negen jaar moet worden gegeven voor de heroplaadbare neurostimulatoren: een volledige garantie voor de eerste vijf jaar en voor de volgende vier jaar een garantie pro rata. Voor de lader 171791-171802 en 171813-171824 is een volledige garantie van negen jaar vereist. De verstrekkingen 171614-171625, 171636-171640 en 171651-171662, moeten ook aan de garantievoorwaarden voldoen.
5. Aantal rechthebbenden
Het aantal rechthebbenden dat voor een tegemoetkoming van de verplichte verzekering in aanmerking kan komen, wordt beperkt tot 25 nieuwe rechthebbenden per jaar.
Zodra het aantal dreigt te worden overschreden, brengt het Secretariaat de Commissie op de hoogte en wordt aan de evaluator gevraagd een verslag te bezorgen. De aard van het verslag wordt vastgelegd door de Commissie.
De Commissie brengt de verplegingsinrichtingen en de verstrekkers van het betrokken hulpmiddel op de hoogte en neemt de noodzakelijke maatregelen.
6. Aanvraagprocedure en formulieren
6.1. Eerste implantatie
De aanvraag tot tegemoetkoming van de verplichte verzekering voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 gebeurt als volgt:
De terugbetalingsaanvraag voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 wordt vóór de implantatie op basis van formulier B-Form-I-07 ingediend door de epileptoloog aan het College van artsen-directeurs.
Die verstrekkingen mogen enkel na akkoord van het College van artsen-directeurs door de verplichte verzekering worden terugbetaald. In geval van twijfel legt het College van artsen-directeurs de aanvraag voor advies voor aan de Commissie Peer Review, die bestaat uit zorgverleners die de revalidatieovereenkomst “referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden” hebben ondertekend. Die Commissie Peer Review moet de aanvraag van het team van het referentiecentrum binnen de drie maanden onderzoeken. De Commissie Peer Review informeert het team van het referentiecentrum dat de aanvraag heeft ingediend, zodat deze de aanvraag kan verdedigen. Tijdens de bespreking van de dossiers die in aanmerking komen voor een tegemoetkoming van de verplichte verzekering voor neurostimulatie kan er altijd een lid van het College van artsen-directeurs of een arts, lid van de Commissie, aanwezig zijn. Minstens twee leden van de Commissie Peer Review die tot twee andere verplegingsinrichtingen dan het aanvragend referentiecentrum behoren, moeten hun akkoord geven.
De Commissie Peer Review bezorgt haar geargumenteerde conclusies (akkoord – weigering – uitstel) aan het College van artsen-directeurs.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging, eventueel op basis van het advies van de Commissie Peer Review, en dit voor elke rechthebbende afzonderlijk.
Het College van artsen-directeurs deelt zijn gemotiveerde beslissing mee binnen de dertig werkdagen. Als dit College een advies van de Commissie Peer Review heeft gevraagd, deelt het zijn gemotiveerde beslissing mee binnen de dertig werkdagen na ontvangst van het verslag van de Commissie Peer Review. Die beslissing wordt aan de epileptoloog van het betrokken referentiecentrum dat tot de overeenkomst is toegetreden, aan de betrokken rechthebbende via zijn verzekeringsinstelling en aan de ziekenhuisapotheker meegedeeld.
De documenten, waaruit blijkt dat voldaan is aan één van de indicaties vermeld onder punt 3, moeten steeds in het medisch dossier van de rechthebbende aanwezig zijn.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171614-171625 worden geattesteerd.
6.2. Vervanging
In geval van hernieuwing van een hulpmiddel dat reeds het voorwerp heeft uitgemaakt van een tegemoetkoming van de verplichte verzekering in het kader van deze overeenkomst, mag de aanvraag voor de verstrekkingen 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 en 171813-171824 na implantatie ingediend worden door de epileptoloog bij het College van artsen-directeurs door middel van het aanvraagformulier B-Form-I-08.
De aanvraag bevat onder andere een medisch evolutieverslag, waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de redenen voor de noodzaak van de hernieuwing zijn opgenomen.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171636-171640 worden geattesteerd.
6.3. Voortijdige vervanging
De procedure beschreven onder punt 6.2. dient te worden toegepast.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171651-171662 worden geattesteerd.
6.4. Derogatie aan de procedure
Voor de rechthebbenden die reeds vóór de inwerkingtreding van deze overeenkomst ingeplant zijn en die vóór de implantatie aan alle voorwaarden bedoeld in punt 3 voldeden, kan een tegemoetkoming van de verplichte verzekering voor de hernieuwing van de neurostimulatoren en toebehoren worden toegekend volgens de modaliteiten voorzien in punt 6.1.
In dat geval bezorgt het team van het referentiecentrum een aanvraagdossier tot tegemoetkoming van de verplichte verzekering voor een hernieuwing aan het College van artsen-directeurs op basis van de formulieren B-Form-I-07 en 08. Dat dossier bevat de documenten van de eerste implantatie waarin wordt aangetoond dat deze implantatie aan de vergoedingscriteria van de verplichte verzekering voldeed en een medisch evolutieverslag waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de rechtvaardiging van de hernieuwing zijn opgenomen.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor de hernieuwing, eventueel op basis van het advies van de Commissie Peer Review.
7. Regels voor attestering
7.1. Cumul en non-cumulregels
Een tegemoetkoming van de verplichte verzekering voor de verstrekking 171496-171500, 171555-171566 of 171614-171625 sluit tijdens een periode van twee jaar een tegemoetkoming van de verzekering voor de verstrekking 170892-170903 uit.
7.2. Andere regels
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 volgen de vergoedingsmodaliteiten van categorie A.
De verstrekkingen 171614-171625, 171636-171640 en 171651-171662 volgen de vergoedingsmodaliteiten van categorie F.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171511-171522 kan pas minstens twee jaar na de verstrekking 171496-171500, 171511-171522 of 171533-171544 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171570-171581 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 of 171651-171662 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171695-171706 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171625, 171636-171640 of 171651-171662 worden toegekend.
7.3. Derogatie van de attesteringsregels
Niet van toepassing.
8. Verbintenissen van de verplegingsinrichting
8.1. De verplegingsinrichting die tot de overeenkomst is toegetreden moet de baseline karakteristieken en de follow-up gegevens van de rechthebbenden die in het kader van deze overeenkomst zijn behandeld nauwgezet bijhouden om de analyse voorzien in punt 9.2. uit te voeren. Deze gegevens moeten in het Elektronisch Patiënten Dossier bewaard worden.
8.2. De inrichtende macht van de verplegingsinrichting is verantwoordelijk voor de onverwijlde mededeling van elke wijziging aan de leidend ambtenaar van de Dienst voor geneeskundige verzorging van het RIZIV en natuurlijk aan alle betrokkenen, waaronder in de eerste plaats de rechthebbenden.
Per e-mail aan het Secretariaat van de Commissie Tegemoetkoming Implantaten en Invasieve Medische Hulpmiddelen, op het volgende adres: implant@riziv-inami.fgov.be.
Wanneer de Leidend ambtenaar nalatigheden vaststelt, brengt hij de verzekeringsinstellingen ervan op de hoogte dat het hulpmiddel voor die verplegingsinrichting niet meer mag worden terugbetaald.
9. Analyse
9.1. De analyse van de gegevens uit deze beperkte klinische toepassing wordt uitgevoerd door de Commissie Peer Review en de verzekeringsinstellingen – verder evaluator genoemd – die mee de overeenkomst ondertekend hebben. Zij verlenen technische en wetenschappelijke ondersteuning en analyseren de resultaten van het medisch hulpmiddel volgens precieze criteria.
9.2. Analyse – Eindverslag
Uiterlijk 6 maanden voor het einde van de overeenkomst moet de evaluator op basis van de verzamelde gegevens een eindverslag opstellen en aan de Commissie bezorgen.
Dit verslag betreft alle rechthebbenden geïmplanteerd met een neurostimulator voor diepe hersenstimulatie in geval van refractaire epilepsie sinds de datum van inwerkingtreding van de terugbetaling.
Dat eindverslag moet minstens de volgende elementen bevatten:
1) het aantal behandelde patiënten, geslacht, leeftijd;
2) analyse van alle inclusie- en exclusiecriteria, waaronder indicaties, contra-indicaties, duur van de symptomen, behandelingen, andere comorbide psychiatrische aandoeningen;
3) resultaten van de neurostimulatie:
a. wijziging van de ernst van de aanvallen
b. wijziging van de aanvalsfrequentie
c. responsratio (= daling met 50 % of meer van de aanvalsfrequentie)
d. wijziging van de medicamenteuze behandeling
4) bijwerkingen, complicaties;
5) een retrospectieve analyse van de directe medische kosten, gerealiseerd met een vergelijking van de cumulatieve kosten gedurende één jaar vóór de implantatie van een DBS-stimulator met de cumulatieve kosten van het jaar na implantatie. De volgende kosten worden bestudeerd:
a. aantal en duur van opnames in verplegingsinrichting en gelinkt aan epilepsie. De technische onderzoeken met betrekking tot diagnostiek of behandeling zijn inbegrepen.
De kosten te wijten aan de prechirurgische evaluatie zijn niet het onderwerp van de studie.
b. aantal bezoeken aan de spoed
c. aantal bezoeken bij de huisarts of de neuroloog
d. aantal en dosis van de anti-epileptische farmaceutische specialiteiten
6) een analyse van de indirecte kosten;
7) een vergelijking van de resultaten met de bestaande literatuur.
De verzekeringsinstellingen zullen de retrospectieve analyse van de directe en indirecte medische kosten zoals gedefinieerd in punten 5 en 6 van de inhoud van het eindverslag bezorgen.
Indien dit verslag niet op de voormelde datum wordt meegedeeld, brengt de Commissie de minister daarvan op de hoogte. Deze kan beslissen om de terugbetaling van het hulpmiddel stop te zetten.
De Commissie zal het eindverslag waarin het hulpmiddel wordt geëvalueerd, als basis kunnen gebruiken voor het opstellen van een definitieve regeling. Die regeling zal door de Commissie aan de Minister worden voorgelegd.
10. Opzeggingsrecht voor elke betrokken partij
De overeenkomst treedt in werking op 1 december 2020 en is geldig tot en met 30 november 2023 maar kan steeds door het RIZIV of door een verplegingsinrichting worden opgezegd met een ter post aangetekende brief die aan de andere partij wordt gericht, mits inachtneming van een opzeggingstermijn van drie maanden die ingaat op de eerste dag van de maand volgend op de datum van verzending van de aangetekende brief.
De overeenkomst verstrijkt zodra de verplegingsinrichting niet meer aan de bepalingen van deze overeenkomst voldoet.
11. Varia
Op verzoek van de Commissie of van de evaluator kan er op elk moment een vergadering worden georganiseerd.
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Afin de pouvoir bénéficier d’une intervention temporaire de l’assurance obligatoire pour les prestations relatives aux neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire, il doit être satisfait aux conditions suivantes :
1. But de la convention
Cette convention a pour but de fixer le remboursement temporaire de l’assurance obligatoire soins de santé ainsi que ses modalités concernant les neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire dans le cadre d’une application clinique limitée pendant la période d’évaluation qui cours du 1er décembre 2020 au 30 novembre 2023 inclus. Pendant cette période, le dispositif sera évalué selon les dispositions prévues au point 9.
2. Critères concernant l’établissement hospitalier
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si elles sont effectuées dans un établissement hospitalier qui répond aux critères suivants et qui a conclu la convention B-ACL-001-bis avec le Comité de l’assurance:
L’établissement hospitalier doit durant la totalité de la durée de la convention répondre aux critères ci-dessous.
2.1. Seuls les établissements hospitaliers ayant signé la convention de rééducation des centres de référence pour bénéficiaires souffrant d’épilepsie rebelle peuvent adhérer à cette convention.
L’établissement hospitalier doit avoir une permanence en neurochirurgie et en neurologie 24 heures sur 24 et 7 jours sur 7.
La pose d’indication, l’évaluation pré-chirurgicale, l’implantation, y compris les remplacements, la rééducation et un suivi à long terme ne peuvent être effectuées que par ces seuls établissements hospitaliers. Le Comité de l'assurance soins de santé dresse, sur proposition du Service des soins de santé, une liste, mise à jour de façon continue, avec la composition de l'équipe par établissement hospitalier et l'envoie pour information à la Commission.
L’établissement hospitalier et les médecins ayant adhéré à la convention B-ACL-001-bis s’engagent à collaborer à l’évaluation visée au point 9.
Le suivi du bénéficiaire au quotidien est permis hors de ces centres de référence.
2.2. Formulaire de candidature pour l’établissement hospitalier
L’établissement hospitalier doit se faire connaître auprès du Service des Soins de santé sur base du formulaire B-Form-II-02
2.3. Accord de coopération
L’établissement hospitalier ne peut conclure ni d’accord de coopération formalisé avec d’autres établissements hospitaliers ni d’associations hospitalières. De plus, l’intervention doit être réalisée dans les murs de l’établissement hospitalier habilité.
3. Critères concernant le bénéficiaire
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le bénéficiaire répond aux critères suivants :
3.1. Critères d’inclusion
1) Le bénéficiaire est atteint d’épilepsie focale avec des crises focales complexes, avec ou sans généralisation secondaire.
2) Le bénéficiaire est atteint d´épilepsie réfractaire, c´est-à-dire qu’un contrôle satisfaisant des crises ne peut pas être obtenu avec un des médicaments antiépileptiques potentiellement efficaces, administré seul ou en combinaison, à des doses thérapeutiques optimales et non associées à des effets secondaires inacceptables, les crises entrainant des incapacités et un handicap.
3) Le bénéficiaire doit avoir été traité par un traitement pharmacologique optimal. Le bénéficiaire n’a pas de contrôle satisfaisant de l’épilepsie avec au minimum 3 thérapies différentes, dont au minimum une association, aux doses optimales et durant une période suffisante pour en apprécier l’efficacité.
4) Le bénéficiaire n’est pas éligible pour une chirurgie ou la chirurgie de l’épilepsie est un échec. L’évaluation préchirurgicale inclut les tests suivants :
a. Enregistrement vidéo-EEG de longue durée avec enregistrement des crises
b. IRM à haute résolution du cerveau
c. FDG-PET du cerveau
d. Évaluation neuropsychologique incluant les éléments suivants :
i. QI
ii. mémoire
iii. fonctions exécutives frontales
e. Évaluation psychiatrique incluant entre autres les éléments suivants :
i. inventaire de dépression de Beck
ii. QoLIE-31
Au cas où un examen ne serait pas réalisable, par exemple suite à un retard mental (qui ne constitue pas en soi une contre-indication à l´implantation d´un stimulateur pour stimulation cérébrale profonde), la raison doit en être clairement mentionnée dans le formulaire.
5) Le bénéficiaire doit être âgé d’au moins 18 ans et de maximum 65 ans lors de l’implantation.
6) Le bénéficiaire est capable d’utiliser un programmateur et de se soumettre aux tests demandés
7) Seuls les bénéficiaires clairement capables, de décider de leur plein gré, via une déclaration de consentement éclairé, de l’implantation d’électrodes et d’un neurostimulateur entrent en ligne de compte. Cette déclaration doit expliquer de manière détaillée les avantages et inconvénients du traitement, les risques ainsi que l’impact psychosocial.
8) Si le bénéficiaire est traité par stimulation du nerf vague et en cas de résultats insuffisants, il faut minimum 2 ans depuis la primo-implantation pour pouvoir implanter un neurostimulateur DBS et inversement.
9) La zone stimulée (cible) est celle qui est reprise dans les indications couvertes par le marquage CE en vigueur.
3.2. Critères d’exclusion
1) Affection neurologique ou médicale grave qui constitue une contre-indication pour une intervention cérébrale.
2) Toute contre-indication chirurgicale pour subir une DBS, y compris les contre-indications connues pour la DBS et/ou pour l’exécution d’une IRM préopératoire, contre-indications dans le cadre d’une intervention sous anesthésie ou autres facteurs à risque pour une intervention chirurgicale (une affection cardio-vasculaire grave, coagulopathie,…).
3) Utilisation inappropriée d’un produit (alcool, drogue,…), abus de substance qui ne permet pas un usage correct de l’appareil ou rendant un suivi médical/psychiatrique systématique impossible.
4) Idées suicidaires
5) Problématique chronique psychotique non stabilisée sous traitement, à l’exception de la psychose péri-ictale
4. Critères concernant le dispositif
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le dispositif répond aux critères suivants:
4.1. Définition
Les dispositifs visés par les prestations 171614-171625, 171636-171640 et 171651-171662 ne portent pas le marquage CE mais doivent faire l'objet d'une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
4.2. Critères
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 et 171695-171706, le dispositif doit répondre aux critères suivants :
Le dispositif doit avoir fait preuve de son efficacité et sa sécurité à l’aide d’ études cliniques. Ces études seront publiées dans une revue « peer-reviewed » reconnue internationalement. Parmi les études cliniques réalisées, il faut au minimum une étude de suivi sur au minimum 100 patients suivis pendant minimum 2 ans, ainsi qu’une étude clinique comparative, randomisée au minimum pendant 3 mois et ayant une puissance statistique déterminée a priori d’au moins 80%. L’étude de suivi et l’étude randomisée peuvent être la même étude.
Un dispositif qui est une adaptation d’un dispositif déjà inscrit sur la liste nominative pour le même distributeur, sans changement du mode d’action et sans impact négatif sur l’efficacité, la sécurité et la qualité, peut être inscrit sans études cliniques à condition que le distributeur décrive les adaptations et leurs conséquences pratiques.
4.3. Conditions de garantie
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 et 171813-171824, le dispositif doit répondre aux conditions de garantie suivantes :
- neurostimulateurs non-rechargeables :
une garantie totale de 24 mois doit être donnée pour les neurostimulateurs non-rechargeables
- neurostimulateurs rechargeables:
une garantie de neuf ans doit être donnée pour les neurostimulateurs rechargeables : une garantie totale pour les cinq premières années et pour les quatre années suivantes une garantie au prorata. Pour le chargeur 171791-171802 et 171813-171824, une garantie totale de neuf ans est exigée. Les prestations 171614-171625, 171636-171640 et 171651-171662 doivent également répondre à ces conditions de garantie.
5. Nombre de bénéficiaires
Le nombre de bénéficiaires qui peuvent entrer en ligne de compte pour une intervention de l’assurance obligatoire est limité à 25 nouveaux bénéficiaires par an.
Dès que le nombre menace d’être dépassé, le Secrétariat en informe la Commission et il est demandé à l’évaluateur de communiquer un rapport. La nature du rapport est établie par la Commission.
La Commission informe les établissements hospitaliers et les distributeurs du dispositif concerné et prend les mesures nécessaires.
6. Procédure de demande et formulaires
6.1 Première implantation
La demande d’intervention de l’assurance obligatoire pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 se déroule comme suit:
La demande d’intervention pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 est introduite avant implantation par l’épileptologue sur base du formulaire B-Form-I-07 au Collège des médecins-directeurs.
Ces prestations ne peuvent faire l’objet d’une intervention de l’assurance qu’après accord du Collège des médecins-directeurs. En cas de doute, le Collège des médecins-directeurs peut transmettre la demande pour avis à la Commission Peer Review, composée des prestataires ayant signé la convention de rééducation « centres de référence pour bénéficiaires souffrant d’épilepsie réfractaire ». Cette Commission Peer Review est alors tenue d’examiner la demande de l’équipe du centre de référence dans les trois mois. La Commission Peer Review avertit l’équipe du centre de référence qui a introduit la demande afin de lui permettre de la défendre. Au cours de la discussion portant sur les dossiers qui entrent en ligne de compte pour une intervention de l’assurance pour la neurostimulation, un membre du Collège des médecins-directeurs ou un médecin, membre de la Commission, peut toujours être présent. Minimum deux membres de la Commission Peer Review issus de deux établissements autres que le centre de référence demandeur doivent donner leur accord.
La Commission Peer Review envoie ses conclusions argumentées (accord-refus-report) au Collège des médecins-directeurs.
Le Collège des médecins-directeurs prend la décision d’une intervention de l’assurance obligatoire, éventuellement sur base de l’avis remis par la Commission Peer Review et ce, pour chaque bénéficiaire individuellement.
Le Collège des médecins-directeurs communique sa décision motivée dans les trente jours ouvrables. Si ce Collège a demandé un avis de la Commission peer-review, il communique sa décision motivée dans les trente jours ouvrables après réception du rapport de la Commission Peer Review. Cette décision est communiquée à l’épileptologue du centre de référence concerné et qui a adhéré à la convention, au bénéficiaire concerné via son organisme assureur et au pharmacien hospitalier.
Les documents desquels il ressort qu'il est satisfait à l’une des indications mentionnées au point 3. doivent être conservés dans le dossier médical du bénéficiaire.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
Une copie de cette dérogation sera fournie au Collège des médecins-directeurs.
Dans ce cas, la prestation 171614-171625 doit être attestée.
6.2. Remplacement
Dans le cas d’un renouvellement d’un dispositif qui a déjà fait l’objet d’une intervention de l’assurance obligatoire dans le cadre de cette convention, la demande pour les prestations 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 et 171813-171824 peut être introduite par l’épileptologue après implantation au Collège des médecins-directeurs au moyen du formulaire de demande B-Form-I-08.
La demande contient notamment un rapport médical de l’évolution, dans lequel doit être mentionné entre autres le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171636-171640 doit être attestée.
6.3 Remplacement anticipé
La procédure expliquée au point 6.2. doit être appliquée.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171651-171662 doit être attestée.
6.4 Dérogation à la procédure
Pour les bénéficiaires qui ont déjà été implantés avant l’entrée en vigueur de la présente convention et qui remplissaient avant implantation toutes les conditions visées au point 3, une intervention de l’assurance obligatoire pour le renouvellement des neurostimulateurs et accessoires peut être accordée suivant les modalités prévues au point 6.1.
Dans ce cas particulier, l’équipe du centre de référence fait parvenir un dossier de demande d’intervention de l’assurance obligatoire pour un renouvellement au Collège des médecins-directeurs sur base des formulaires B-Form-I-07 et 08. Ce dossier comprendra les documents de la première implantation démontrant que cette implantation répondait aux critères d’intervention de l’assurance obligatoire ainsi qu’un rapport médical de l’évolution, dans lequel doit être entre autres mentionné le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Le Collège prendra la décision d’une intervention de l’assurance obligatoire pour le renouvellement, éventuellement sur base de l’avis de la Commission Peer Review.
7. Règles d’attestation
7.1 Règles de cumul et de non-cumul
Une intervention de l'assurance obligatoire pour la prestation 171496-171500, 171555-171566 ou 171614-171625 exclut, pendant une période de deux ans, une intervention de l'assurance pour la prestation 170892-170903.
7.2. Autres règles
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 suivent les modalités de remboursement de la catégorie A.
Les prestations 171614-171625, 171636-171640 ou 171651-171662 suivent les modalités de remboursement de la catégorie F.
L’intervention de l’assurance obligatoire pour la prestation 171511-171522 ne peut être accordée que minimum deux ans après la prestation 171496-171500, 171511-171522 ou 171533-171544.
L’intervention de l’assurance obligatoire pour la prestation 171570-171581 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 ou 171651-171662.
L’intervention de l’assurance obligatoire pour la prestation 171636-171640 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603,171614-171625, 171636-171640 ou 171651-171662..
7.3 Dérogation aux règles d’attestation
Pas d’application.
8. Engagements de l’établissement hospitalier
8.1. L’établissement hospitalier qui a adhéré à la convention est tenu de tenir à jour consciencieusement les caractéristiques de référence (baseline) et les données de suivi des bénéficiaires traités dans le cadre de cette convention afin de réaliser l'analyse prévue au point 9.2. Ces données doivent être conservées dans le Dossier Patient Informatisé.
8.2. Le pouvoir organisateur de l'établissement hospitalier porte la responsabilité de communiquer, sans délai, chaque modification au Fonctionnaire dirigeant du Service des soins de santé de l'INAMI et bien entendu à tous ceux qui sont concernés, dont en premier lieu les bénéficiaires.
Par e-mail au Secrétariat de la Commission de remboursement des implants et des dispositifs médicaux invasifs, à l’adresse suivante : implant@riziv-inami.fgov.be.
Lorsque le Fonctionnaire dirigeant constate des manquements, il informe les organismes assureurs du fait que le dispositif n’est plus remboursable pour cet établissement hospitalier.
9. Analyse
9.1. L’analyse des données émanant de cette application clinique limitée est effectuée par la Commission Peer Review et les organismes assureurs – nommés également évaluateur – qui ont signé la convention. Ils apportent un support technique et scientifique et évaluent les résultats du dispositif médical selon des critères précis.
9.2. Analyse – Rapport final
Au plus tard 6 mois avant la fin de la convention, l’évaluateur doit rédiger un rapport final sur base des données collectées et le transmettre à la Commission.
Ce rapport concerne tous les bénéficiaires implantés avec un neurostimulateur pour stimulation cérébrale profonde en cas d’épilepsie réfractaire depuis la date d’entrée en vigueur du remboursement.
Ce rapport final comprend au minimum les éléments suivants :
1) le nombre de patients traités, sexe, âge ;
2) analyse de tous les critères d’inclusion et d’exclusion dont indications, contre-indications, durée des symptômes, traitements, autres affections psychiatriques comorbides ;
3) résultats de la neurostimulation :
a. modification de la sévérité des crises
b. modification de la fréquence des crises
c. taux de répondeurs (= diminution de 50% ou plus de la fréquence des crises)
d. modification du traitement médicamenteux
4) effets secondaires, complications
5) une analyse rétrospective des coûts médicaux directs réalisée avec une comparaison des coûts cumulatifs pendant une période de un an avant l’implantation du stimulateur DBS avec les coûts cumulatifs de l’année suivant l’implantation. Les coûts étudiés seront les suivants :
a. nombre et durée des admissions en établissement hospitalier et liées à l’épilepsie. Les examens techniques relatifs au diagnostic et au traitement sont inclus, les coûts
dus à l’évaluation préchirurgicale ne font pas l’objet de l’étude
b. nombre de visite aux urgences
c. nombre de visite chez le médecin généraliste ou le neurologue
d. nombre et dose de spécialités pharmaceutiques antiépileptiques
6) une analyse des coûts indirects
7) une comparaison des résultats avec la littérature existante
Les organismes assureurs fourniront l’analyse rétrospective des coûts médicaux directs et indirects comme défini aux points 5 et 6 du contenu du rapport final.
Si ce rapport n’est pas communiqué à la date mentionnée ci-dessus, la Commission en informe le Ministre. Celui-ci peut prendre la décision de suspendre le remboursement du dispositif.
La Commission utilisera le rapport final qui évalue le dispositif comme base pour rédiger un règlement définitif. Ce règlement sera soumis au Ministre par l’intermédiaire de la Commission.
10. Droit de résiliation pour chaque partie prenante
La convention entre en vigueur le 1er décembre 2020 et est valable jusqu’au 30 novembre 2023 inclus mais peut toujours être résiliée par l’INAMI ou par un établissement hospitalier par lettre recommandée à la poste, adressée à l'autre partie, en respectant le délai de résiliation de trois mois qui prend cours le premier jour du mois suivant la date d'envoi de la lettre recommandée.
La convention expire dès que l’établissement hospitalier ne répond plus aux dispositions de cette convention.
11. Divers
A la demande de la Commission ou de l’évaluateur, une réunion peut être organisée à tout moment.
Teneinde een tijdelijke tegemoetkoming van de verplichte verzekering te kunnen genieten voor de verstrekkingen betreffende de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie moet aan volgende voorwaarden worden voldaan:
1. Doel van de overeenkomst
Deze overeenkomst heeft tot doel de tijdelijke tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging inzake de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie alsook de modaliteiten ervan te bepalen in het kader van een beperkte klinische toepassing gedurende de evaluatieperiode die loopt van 1 december 2020 tot en met 30 november 2023. Gedurende die periode wordt het hulpmiddel geëvalueerd volgens de bepalingen voorzien in punt 9.
2. Criteria betreffende de verplegingsinrichting
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien ze zijn uitgevoerd in een verplegingsinrichting die aan de volgende criteria voldoet en die de overeenkomst B-BKT-001-bis heeft afgesloten met het Verzekeringscomité:
De verplegingsinrichting moet gedurende de volledige looptijd van de overeenkomst voldoen aan de onderstaande criteria.
2.1. Enkel de verplegingsinrichtingen die de revalidatieovereenkomst van de referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden, hebben ondertekend, kunnen toetreden tot deze overeenkomst.
De verplegingsinrichting moet 24 uur op 24 en 7 dagen op 7 een neurochirurgische en neurologische permanentie hebben.
De indicatiestelling, de prechirurgische evaluatie, de implantatie, met inbegrip van de vervangingen, de revalidatie en een follow-up op lange termijn mogen enkel door deze verplegingsinrichtingen worden uitgevoerd. Het Comité van de verzekering voor geneeskundige verzorging stelt, op voorstel van de Dienst voor geneeskundige verzorging, een lijst op die continu wordt bijgewerkt, met vermelding van de samenstelling van het team per verplegingsinrichting en bezorgt die lijst ter informatie aan de Commissie.
De verplegingsinrichting en de artsen toegetreden tot de overeenkomst B-BKT-001-bis engageren zich tot het meewerken aan de evaluatie zoals bedoeld in punt 9.
De dagelijkse follow-up van de rechthebbende mag buiten die referentiecentra worden verricht.
2.2. Kandidatuurformulier voor de verplegingsinrichting
De verplegingsinrichting moet zich kenbaar maken bij de Dienst voor Geneeskundige verzorging op basis van het formulier B-Form-II-02.
2.3. Samenwerkingsakkoord
De verplegingsinrichting mag geen geformaliseerd samenwerkingsakkoord met andere verplegingsinrichtingen of met andere ziekenhuisverenigingen sluiten. Bovendien moet de ingreep binnen de muren van het bevoegde verplegingsinrichting worden uitgevoerd.
3. Criteria betreffende de rechthebbende
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien de rechthebbende aan de volgende criteria voldoet:
3.1. Inclusiecriteria
1) De rechthebbende lijdt aan focale epilepsie met focale complexe aanvallen met of zonder secundaire generalisering.
2) De rechthebbende lijdt aan refractaire epilepsie, d.w.z. waarbij de aanvallen onvoldoende onder controle kunnen worden gehouden met één van de potentieel doeltreffende anti-epileptica, die alleen of in combinatie worden toegediend, met optimale therapeutische dosissen waaraan geen onaanvaardbare bijwerkingen zijn verbonden; de aanvallen kunnen bovendien arbeidsongeschiktheid of een handicap tot gevolg hebben.
3) De rechthebbende moet een optimale farmacologische behandeling hebben gekregen. De rechthebbende is niet in staat om de epilepsie voldoende onder controle te houden met minstens 3 verschillende therapieën, waarvan minstens één combinatie, met optimale dosissen en gedurende een periode die volstaat om de doeltreffendheid ervan te beoordelen.
4) De rechthebbende komt niet in aanmerking voor chirurgie of de epilepsiechirurgie is een mislukking. De prechirurgische evaluatie omvat de volgende tests:
a. langdurige EEG-videomonitoring met registratie van de aanvallen
b. hogeresolutie-MRI van de hersenen
c. FDG-PET-scan van de hersenen
d. neuropsychologische evaluatie die de volgende elementen bevat:
i. IQ
ii. geheugenfuncties
iii.
e. psychiatrische evaluatie die onder andere de volgende elementen bevat:
i. Beck Depression Inventory
ii. QOLIE-31
In geval dit onderzoek niet gerealiseerd kan worden, bijvoorbeeld als gevolg van een mentale achterstand (die op zich geen tegenindicatie vormt voor de implantatie van een stimulator voor diepe hersenstimulatie), dient de reden duidelijk te worden vermeld op het formulier.
5) De rechthebbende moet op het moment van de implantatie minstens 18 jaar en maximaal 65 jaar oud zijn.
6) De rechthebbende kan een patiëntenprogrameerapparaat gebruiken en is bereid om zich aan de gevraagde tests te onderwerpen.
7) Alleen de rechthebbenden die duidelijk in staat zijn om, uit eigen wil, via een informed consent over de implantatie van elektroden en een neurostimulator te beslissen, komen in aanmerking. In die verklaring moeten omstandig de voor- en nadelen van de behandeling, de risico’s en de psychosociale impact worden toegelicht.
8) Als de rechthebbende met nervus vagusstimulatie wordt behandeld en indien daarmee onvoldoende resultaten worden bereikt, moet de rechthebbende vanaf de primo-implantatie minstens 2 jaar op de implantatie van een DBS-neurostimulator wachten en vice versa.
9) De gestimuleerde zone (doelgebied) is de zone die in de indicaties wordt vermeld en die door de huidige CE-markering wordt gedekt.
3.2. Exclusiecriteria
1) Ernstige neurologische of medische aandoening die een contra-indicatie is voor een cerebrale ingreep.
2) Elke chirurgische contra-indicatie om DBS te ondergaan, met inbegrip van de contra-indicaties die voor de DBS en/of voor de uitvoering van een preoperatieve MRI bekend zijn, contra-indicaties in het kader van een ingreep onder anesthesie of andere risicofactoraren voor een chirurgische ingreep (een ernstige cardiovasculaire aandoening, coagulopathie, …).
3) Ongepast gebruik van een product (alcohol, drugs, …)/middelenmisbruik waardoor het toestel niet correct kan worden gebruikt of waardoor er geen systematische medische/psychiatrische follow-up mogelijk is.
4) Suïcidale gedachten
5) Chronische psychotische problematiek die niet gestabiliseerd is onder behandeling, met uitzondering van een peri-ictale psychose
4. Criteria betreffende het hulpmiddel
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien het hulpmiddel aan de volgende criteria voldoet:
4.1. Definitie
De hulpmiddelen bedoeld onder de verstrekkingen 171614-171625, 171636-171640 en 171651-171662 dragen geen CE-markering, maar moeten het voorwerp uitmaken van een derogatie toegekend door de Minister die bevoegd is voor Volksgezondheid.
4.2. Criteria
Om te kunnen worden opgenomen op de nominatieve lijst voor de 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 en 171695-171706 moet het hulpmiddel beantwoorden aan de volgende criteria:
De werkzaamheid en veiligheid van het hulpmiddel moeten zijn aangetoond met behulp van klinische studies. Deze studies dienen gepubliceerd te zijn in een internationaal erkend peer reviewed tijdschrift. Onder de gerealiseerde klinische studies, moet er minstens een follow-up studie zijn met minimum 100 patiënten die gedurende minimaal 2 jaar gevolgd worden, evenals een vergelijkende, gerandomiseerde klinische studie gedurende minimum 3 maand en met een a priori vastgelegde statistische power van minstens 80%. De follow-up studie en de gerandomiseerde studie mogen dezelfde studie zijn.
Een hulpmiddel dat een aanpassing is van een hulpmiddel dat reeds op de nominatieve lijst voor dezelfde verdeler ingeschreven is, zonder wijziging van het werkingsmechanisme en zonder negatieve impact op de werkzaamheid, de veiligheid en de kwaliteit, kan ingeschreven worden zonder klinische studies op voorwaarde dat de verdeler de aanpassingen en hun praktische consequenties beschrijft.
4.3. Garantievoorwaarden
Om te kunnen worden opgenomen op de nominatieve lijst voor de verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 en 171813-171824 moet het hulpmiddel beantwoorden aan de volgende garantievoorwaarden:
- Niet heroplaadbare neurostimulatoren:
een totale garantie van 24 maanden moet worden gegeven voor de niet heroplaadbare neurostimulatoren
- Heroplaadbare neurostimulatoren:
een garantie van negen jaar moet worden gegeven voor de heroplaadbare neurostimulatoren: een volledige garantie voor de eerste vijf jaar en voor de volgende vier jaar een garantie pro rata. Voor de lader 171791-171802 en 171813-171824 is een volledige garantie van negen jaar vereist. De verstrekkingen 171614-171625, 171636-171640 en 171651-171662, moeten ook aan de garantievoorwaarden voldoen.
5. Aantal rechthebbenden
Het aantal rechthebbenden dat voor een tegemoetkoming van de verplichte verzekering in aanmerking kan komen, wordt beperkt tot 25 nieuwe rechthebbenden per jaar.
Zodra het aantal dreigt te worden overschreden, brengt het Secretariaat de Commissie op de hoogte en wordt aan de evaluator gevraagd een verslag te bezorgen. De aard van het verslag wordt vastgelegd door de Commissie.
De Commissie brengt de verplegingsinrichtingen en de verstrekkers van het betrokken hulpmiddel op de hoogte en neemt de noodzakelijke maatregelen.
6. Aanvraagprocedure en formulieren
6.1. Eerste implantatie
De aanvraag tot tegemoetkoming van de verplichte verzekering voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 gebeurt als volgt:
De terugbetalingsaanvraag voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 wordt vóór de implantatie op basis van formulier B-Form-I-07 ingediend door de epileptoloog aan het College van artsen-directeurs.
Die verstrekkingen mogen enkel na akkoord van het College van artsen-directeurs door de verplichte verzekering worden terugbetaald. In geval van twijfel legt het College van artsen-directeurs de aanvraag voor advies voor aan de Commissie Peer Review, die bestaat uit zorgverleners die de revalidatieovereenkomst “referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden” hebben ondertekend. Die Commissie Peer Review moet de aanvraag van het team van het referentiecentrum binnen de drie maanden onderzoeken. De Commissie Peer Review informeert het team van het referentiecentrum dat de aanvraag heeft ingediend, zodat deze de aanvraag kan verdedigen. Tijdens de bespreking van de dossiers die in aanmerking komen voor een tegemoetkoming van de verplichte verzekering voor neurostimulatie kan er altijd een lid van het College van artsen-directeurs of een arts, lid van de Commissie, aanwezig zijn. Minstens twee leden van de Commissie Peer Review die tot twee andere verplegingsinrichtingen dan het aanvragend referentiecentrum behoren, moeten hun akkoord geven.
De Commissie Peer Review bezorgt haar geargumenteerde conclusies (akkoord – weigering – uitstel) aan het College van artsen-directeurs.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging, eventueel op basis van het advies van de Commissie Peer Review, en dit voor elke rechthebbende afzonderlijk.
Het College van artsen-directeurs deelt zijn gemotiveerde beslissing mee binnen de dertig werkdagen. Als dit College een advies van de Commissie Peer Review heeft gevraagd, deelt het zijn gemotiveerde beslissing mee binnen de dertig werkdagen na ontvangst van het verslag van de Commissie Peer Review. Die beslissing wordt aan de epileptoloog van het betrokken referentiecentrum dat tot de overeenkomst is toegetreden, aan de betrokken rechthebbende via zijn verzekeringsinstelling en aan de ziekenhuisapotheker meegedeeld.
De documenten, waaruit blijkt dat voldaan is aan één van de indicaties vermeld onder punt 3, moeten steeds in het medisch dossier van de rechthebbende aanwezig zijn.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171614-171625 worden geattesteerd.
6.2. Vervanging
In geval van hernieuwing van een hulpmiddel dat reeds het voorwerp heeft uitgemaakt van een tegemoetkoming van de verplichte verzekering in het kader van deze overeenkomst, mag de aanvraag voor de verstrekkingen 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 en 171813-171824 na implantatie ingediend worden door de epileptoloog bij het College van artsen-directeurs door middel van het aanvraagformulier B-Form-I-08.
De aanvraag bevat onder andere een medisch evolutieverslag, waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de redenen voor de noodzaak van de hernieuwing zijn opgenomen.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171636-171640 worden geattesteerd.
6.3. Voortijdige vervanging
De procedure beschreven onder punt 6.2. dient te worden toegepast.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171651-171662 worden geattesteerd.
6.4. Derogatie aan de procedure
Voor de rechthebbenden die reeds vóór de inwerkingtreding van deze overeenkomst ingeplant zijn en die vóór de implantatie aan alle voorwaarden bedoeld in punt 3 voldeden, kan een tegemoetkoming van de verplichte verzekering voor de hernieuwing van de neurostimulatoren en toebehoren worden toegekend volgens de modaliteiten voorzien in punt 6.1.
In dat geval bezorgt het team van het referentiecentrum een aanvraagdossier tot tegemoetkoming van de verplichte verzekering voor een hernieuwing aan het College van artsen-directeurs op basis van de formulieren B-Form-I-07 en 08. Dat dossier bevat de documenten van de eerste implantatie waarin wordt aangetoond dat deze implantatie aan de vergoedingscriteria van de verplichte verzekering voldeed en een medisch evolutieverslag waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de rechtvaardiging van de hernieuwing zijn opgenomen.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor de hernieuwing, eventueel op basis van het advies van de Commissie Peer Review.
7. Regels voor attestering
7.1. Cumul en non-cumulregels
Een tegemoetkoming van de verplichte verzekering voor de verstrekking 171496-171500, 171555-171566 of 171614-171625 sluit tijdens een periode van twee jaar een tegemoetkoming van de verzekering voor de verstrekking 170892-170903 uit.
7.2. Andere regels
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 volgen de vergoedingsmodaliteiten van categorie A.
De verstrekkingen 171614-171625, 171636-171640 en 171651-171662 volgen de vergoedingsmodaliteiten van categorie F.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171511-171522 kan pas minstens twee jaar na de verstrekking 171496-171500, 171511-171522 of 171533-171544 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171570-171581 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 of 171651-171662 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171695-171706 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171625, 171636-171640 of 171651-171662 worden toegekend.
7.3. Derogatie van de attesteringsregels
Niet van toepassing.
8. Verbintenissen van de verplegingsinrichting
8.1. De verplegingsinrichting die tot de overeenkomst is toegetreden moet de baseline karakteristieken en de follow-up gegevens van de rechthebbenden die in het kader van deze overeenkomst zijn behandeld nauwgezet bijhouden om de analyse voorzien in punt 9.2. uit te voeren. Deze gegevens moeten in het Elektronisch Patiënten Dossier bewaard worden.
8.2. De inrichtende macht van de verplegingsinrichting is verantwoordelijk voor de onverwijlde mededeling van elke wijziging aan de leidend ambtenaar van de Dienst voor geneeskundige verzorging van het RIZIV en natuurlijk aan alle betrokkenen, waaronder in de eerste plaats de rechthebbenden.
Per e-mail aan het Secretariaat van de Commissie Tegemoetkoming Implantaten en Invasieve Medische Hulpmiddelen, op het volgende adres: implant@riziv-inami.fgov.be.
Wanneer de Leidend ambtenaar nalatigheden vaststelt, brengt hij de verzekeringsinstellingen ervan op de hoogte dat het hulpmiddel voor die verplegingsinrichting niet meer mag worden terugbetaald.
9. Analyse
9.1. De analyse van de gegevens uit deze beperkte klinische toepassing wordt uitgevoerd door de Commissie Peer Review en de verzekeringsinstellingen – verder evaluator genoemd – die mee de overeenkomst ondertekend hebben. Zij verlenen technische en wetenschappelijke ondersteuning en analyseren de resultaten van het medisch hulpmiddel volgens precieze criteria.
9.2. Analyse – Eindverslag
Uiterlijk 6 maanden voor het einde van de overeenkomst moet de evaluator op basis van de verzamelde gegevens een eindverslag opstellen en aan de Commissie bezorgen.
Dit verslag betreft alle rechthebbenden geïmplanteerd met een neurostimulator voor diepe hersenstimulatie in geval van refractaire epilepsie sinds de datum van inwerkingtreding van de terugbetaling.
Dat eindverslag moet minstens de volgende elementen bevatten:
1) het aantal behandelde patiënten, geslacht, leeftijd;
2) analyse van alle inclusie- en exclusiecriteria, waaronder indicaties, contra-indicaties, duur van de symptomen, behandelingen, andere comorbide psychiatrische aandoeningen;
3) resultaten van de neurostimulatie:
a. wijziging van de ernst van de aanvallen
b. wijziging van de aanvalsfrequentie
c. responsratio (= daling met 50 % of meer van de aanvalsfrequentie)
d. wijziging van de medicamenteuze behandeling
4) bijwerkingen, complicaties;
5) een retrospectieve analyse van de directe medische kosten, gerealiseerd met een vergelijking van de cumulatieve kosten gedurende één jaar vóór de implantatie van een DBS-stimulator met de cumulatieve kosten van het jaar na implantatie. De volgende kosten worden bestudeerd:
a. aantal en duur van opnames in verplegingsinrichting en gelinkt aan epilepsie. De technische onderzoeken met betrekking tot diagnostiek of behandeling zijn inbegrepen.
De kosten te wijten aan de prechirurgische evaluatie zijn niet het onderwerp van de studie.
b. aantal bezoeken aan de spoed
c. aantal bezoeken bij de huisarts of de neuroloog
d. aantal en dosis van de anti-epileptische farmaceutische specialiteiten
6) een analyse van de indirecte kosten;
7) een vergelijking van de resultaten met de bestaande literatuur.
De verzekeringsinstellingen zullen de retrospectieve analyse van de directe en indirecte medische kosten zoals gedefinieerd in punten 5 en 6 van de inhoud van het eindverslag bezorgen.
Indien dit verslag niet op de voormelde datum wordt meegedeeld, brengt de Commissie de minister daarvan op de hoogte. Deze kan beslissen om de terugbetaling van het hulpmiddel stop te zetten.
De Commissie zal het eindverslag waarin het hulpmiddel wordt geëvalueerd, als basis kunnen gebruiken voor het opstellen van een definitieve regeling. Die regeling zal door de Commissie aan de Minister worden voorgelegd.
10. Opzeggingsrecht voor elke betrokken partij
De overeenkomst treedt in werking op 1 december 2020 en is geldig tot en met 30 november 2023 maar kan steeds door het RIZIV of door een verplegingsinrichting worden opgezegd met een ter post aangetekende brief die aan de andere partij wordt gericht, mits inachtneming van een opzeggingstermijn van drie maanden die ingaat op de eerste dag van de maand volgend op de datum van verzending van de aangetekende brief.
De overeenkomst verstrijkt zodra de verplegingsinrichting niet meer aan de bepalingen van deze overeenkomst voldoet.
11. Varia
Op verzoek van de Commissie of van de evaluator kan er op elk moment een vergadering worden georganiseerd.
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Afin de pouvoir bénéficier d’une intervention temporaire de l’assurance obligatoire pour les prestations relatives aux neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire, il doit être satisfait aux conditions suivantes :
1. But de la convention
Cette convention a pour but de fixer le remboursement temporaire de l’assurance obligatoire soins de santé ainsi que ses modalités concernant les neurostimulateurs et accessoires pour stimulation cérébrale profonde en cas d’épilepsie réfractaire dans le cadre d’une application clinique limitée pendant la période d’évaluation qui cours du 1er décembre 2020 au 30 novembre 2023 inclus. Pendant cette période, le dispositif sera évalué selon les dispositions prévues au point 9.
2. Critères concernant l’établissement hospitalier
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si elles sont effectuées dans un établissement hospitalier qui répond aux critères suivants et qui a conclu la convention B-ACL-001-bis avec le Comité de l’assurance:
L’établissement hospitalier doit durant la totalité de la durée de la convention répondre aux critères ci-dessous.
2.1. Seuls les établissements hospitaliers ayant signé la convention de rééducation des centres de référence pour bénéficiaires souffrant d’épilepsie rebelle peuvent adhérer à cette convention.
L’établissement hospitalier doit avoir une permanence en neurochirurgie et en neurologie 24 heures sur 24 et 7 jours sur 7.
La pose d’indication, l’évaluation pré-chirurgicale, l’implantation, y compris les remplacements, la rééducation et un suivi à long terme ne peuvent être effectuées que par ces seuls établissements hospitaliers. Le Comité de l'assurance soins de santé dresse, sur proposition du Service des soins de santé, une liste, mise à jour de façon continue, avec la composition de l'équipe par établissement hospitalier et l'envoie pour information à la Commission.
L’établissement hospitalier et les médecins ayant adhéré à la convention B-ACL-001-bis s’engagent à collaborer à l’évaluation visée au point 9.
Le suivi du bénéficiaire au quotidien est permis hors de ces centres de référence.
2.2. Formulaire de candidature pour l’établissement hospitalier
L’établissement hospitalier doit se faire connaître auprès du Service des Soins de santé sur base du formulaire B-Form-II-02
2.3. Accord de coopération
L’établissement hospitalier ne peut conclure ni d’accord de coopération formalisé avec d’autres établissements hospitaliers ni d’associations hospitalières. De plus, l’intervention doit être réalisée dans les murs de l’établissement hospitalier habilité.
3. Critères concernant le bénéficiaire
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le bénéficiaire répond aux critères suivants :
3.1. Critères d’inclusion
1) Le bénéficiaire est atteint d’épilepsie focale avec des crises focales complexes, avec ou sans généralisation secondaire.
2) Le bénéficiaire est atteint d´épilepsie réfractaire, c´est-à-dire qu’un contrôle satisfaisant des crises ne peut pas être obtenu avec un des médicaments antiépileptiques potentiellement efficaces, administré seul ou en combinaison, à des doses thérapeutiques optimales et non associées à des effets secondaires inacceptables, les crises entrainant des incapacités et un handicap.
3) Le bénéficiaire doit avoir été traité par un traitement pharmacologique optimal. Le bénéficiaire n’a pas de contrôle satisfaisant de l’épilepsie avec au minimum 3 thérapies différentes, dont au minimum une association, aux doses optimales et durant une période suffisante pour en apprécier l’efficacité.
4) Le bénéficiaire n’est pas éligible pour une chirurgie ou la chirurgie de l’épilepsie est un échec. L’évaluation préchirurgicale inclut les tests suivants :
a. Enregistrement vidéo-EEG de longue durée avec enregistrement des crises
b. IRM à haute résolution du cerveau
c. FDG-PET du cerveau
d. Évaluation neuropsychologique incluant les éléments suivants :
i. QI
ii. mémoire
iii. fonctions exécutives frontales
e. Évaluation psychiatrique incluant entre autres les éléments suivants :
i. inventaire de dépression de Beck
ii. QoLIE-31
Au cas où un examen ne serait pas réalisable, par exemple suite à un retard mental (qui ne constitue pas en soi une contre-indication à l´implantation d´un stimulateur pour stimulation cérébrale profonde), la raison doit en être clairement mentionnée dans le formulaire.
5) Le bénéficiaire doit être âgé d’au moins 18 ans et de maximum 65 ans lors de l’implantation.
6) Le bénéficiaire est capable d’utiliser un programmateur et de se soumettre aux tests demandés
7) Seuls les bénéficiaires clairement capables, de décider de leur plein gré, via une déclaration de consentement éclairé, de l’implantation d’électrodes et d’un neurostimulateur entrent en ligne de compte. Cette déclaration doit expliquer de manière détaillée les avantages et inconvénients du traitement, les risques ainsi que l’impact psychosocial.
8) Si le bénéficiaire est traité par stimulation du nerf vague et en cas de résultats insuffisants, il faut minimum 2 ans depuis la primo-implantation pour pouvoir implanter un neurostimulateur DBS et inversement.
9) La zone stimulée (cible) est celle qui est reprise dans les indications couvertes par le marquage CE en vigueur.
3.2. Critères d’exclusion
1) Affection neurologique ou médicale grave qui constitue une contre-indication pour une intervention cérébrale.
2) Toute contre-indication chirurgicale pour subir une DBS, y compris les contre-indications connues pour la DBS et/ou pour l’exécution d’une IRM préopératoire, contre-indications dans le cadre d’une intervention sous anesthésie ou autres facteurs à risque pour une intervention chirurgicale (une affection cardio-vasculaire grave, coagulopathie,…).
3) Utilisation inappropriée d’un produit (alcool, drogue,…), abus de substance qui ne permet pas un usage correct de l’appareil ou rendant un suivi médical/psychiatrique systématique impossible.
4) Idées suicidaires
5) Problématique chronique psychotique non stabilisée sous traitement, à l’exception de la psychose péri-ictale
4. Critères concernant le dispositif
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 ne peuvent faire l’objet d’une intervention de l’assurance obligatoire que si le dispositif répond aux critères suivants:
4.1. Définition
Les dispositifs visés par les prestations 171614-171625, 171636-171640 et 171651-171662 ne portent pas le marquage CE mais doivent faire l'objet d'une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
4.2. Critères
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 et 171695-171706, le dispositif doit répondre aux critères suivants :
Le dispositif doit avoir fait preuve de son efficacité et sa sécurité à l’aide d’ études cliniques. Ces études seront publiées dans une revue « peer-reviewed » reconnue internationalement. Parmi les études cliniques réalisées, il faut au minimum une étude de suivi sur au minimum 100 patients suivis pendant minimum 2 ans, ainsi qu’une étude clinique comparative, randomisée au minimum pendant 3 mois et ayant une puissance statistique déterminée a priori d’au moins 80%. L’étude de suivi et l’étude randomisée peuvent être la même étude.
Un dispositif qui est une adaptation d’un dispositif déjà inscrit sur la liste nominative pour le même distributeur, sans changement du mode d’action et sans impact négatif sur l’efficacité, la sécurité et la qualité, peut être inscrit sans études cliniques à condition que le distributeur décrive les adaptations et leurs conséquences pratiques.
4.3. Conditions de garantie
Afin de pouvoir être repris sur la liste nominative pour les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 et 171813-171824, le dispositif doit répondre aux conditions de garantie suivantes :
- neurostimulateurs non-rechargeables :
une garantie totale de 24 mois doit être donnée pour les neurostimulateurs non-rechargeables
- neurostimulateurs rechargeables:
une garantie de neuf ans doit être donnée pour les neurostimulateurs rechargeables : une garantie totale pour les cinq premières années et pour les quatre années suivantes une garantie au prorata. Pour le chargeur 171791-171802 et 171813-171824, une garantie totale de neuf ans est exigée. Les prestations 171614-171625, 171636-171640 et 171651-171662 doivent également répondre à ces conditions de garantie.
5. Nombre de bénéficiaires
Le nombre de bénéficiaires qui peuvent entrer en ligne de compte pour une intervention de l’assurance obligatoire est limité à 25 nouveaux bénéficiaires par an.
Dès que le nombre menace d’être dépassé, le Secrétariat en informe la Commission et il est demandé à l’évaluateur de communiquer un rapport. La nature du rapport est établie par la Commission.
La Commission informe les établissements hospitaliers et les distributeurs du dispositif concerné et prend les mesures nécessaires.
6. Procédure de demande et formulaires
6.1 Première implantation
La demande d’intervention de l’assurance obligatoire pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 se déroule comme suit:
La demande d’intervention pour les prestations 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 et 171791-171802 est introduite avant implantation par l’épileptologue sur base du formulaire B-Form-I-07 au Collège des médecins-directeurs.
Ces prestations ne peuvent faire l’objet d’une intervention de l’assurance qu’après accord du Collège des médecins-directeurs. En cas de doute, le Collège des médecins-directeurs peut transmettre la demande pour avis à la Commission Peer Review, composée des prestataires ayant signé la convention de rééducation « centres de référence pour bénéficiaires souffrant d’épilepsie réfractaire ». Cette Commission Peer Review est alors tenue d’examiner la demande de l’équipe du centre de référence dans les trois mois. La Commission Peer Review avertit l’équipe du centre de référence qui a introduit la demande afin de lui permettre de la défendre. Au cours de la discussion portant sur les dossiers qui entrent en ligne de compte pour une intervention de l’assurance pour la neurostimulation, un membre du Collège des médecins-directeurs ou un médecin, membre de la Commission, peut toujours être présent. Minimum deux membres de la Commission Peer Review issus de deux établissements autres que le centre de référence demandeur doivent donner leur accord.
La Commission Peer Review envoie ses conclusions argumentées (accord-refus-report) au Collège des médecins-directeurs.
Le Collège des médecins-directeurs prend la décision d’une intervention de l’assurance obligatoire, éventuellement sur base de l’avis remis par la Commission Peer Review et ce, pour chaque bénéficiaire individuellement.
Le Collège des médecins-directeurs communique sa décision motivée dans les trente jours ouvrables. Si ce Collège a demandé un avis de la Commission peer-review, il communique sa décision motivée dans les trente jours ouvrables après réception du rapport de la Commission Peer Review. Cette décision est communiquée à l’épileptologue du centre de référence concerné et qui a adhéré à la convention, au bénéficiaire concerné via son organisme assureur et au pharmacien hospitalier.
Les documents desquels il ressort qu'il est satisfait à l’une des indications mentionnées au point 3. doivent être conservés dans le dossier médical du bénéficiaire.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions.
Une copie de cette dérogation sera fournie au Collège des médecins-directeurs.
Dans ce cas, la prestation 171614-171625 doit être attestée.
6.2. Remplacement
Dans le cas d’un renouvellement d’un dispositif qui a déjà fait l’objet d’une intervention de l’assurance obligatoire dans le cadre de cette convention, la demande pour les prestations 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 et 171813-171824 peut être introduite par l’épileptologue après implantation au Collège des médecins-directeurs au moyen du formulaire de demande B-Form-I-08.
La demande contient notamment un rapport médical de l’évolution, dans lequel doit être mentionné entre autres le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171636-171640 doit être attestée.
6.3 Remplacement anticipé
La procédure expliquée au point 6.2. doit être appliquée.
Jusqu’à l’obtention d’un marquage CE, les neurostimulateurs rechargeables ne peuvent être implantés qu’après avoir obtenu une dérogation accordée par le Ministre ayant la Santé publique dans ses attributions. Une copie de cette dérogation sera fournie au collège des médecins-directeurs.
Dans ce cas, la prestation 171651-171662 doit être attestée.
6.4 Dérogation à la procédure
Pour les bénéficiaires qui ont déjà été implantés avant l’entrée en vigueur de la présente convention et qui remplissaient avant implantation toutes les conditions visées au point 3, une intervention de l’assurance obligatoire pour le renouvellement des neurostimulateurs et accessoires peut être accordée suivant les modalités prévues au point 6.1.
Dans ce cas particulier, l’équipe du centre de référence fait parvenir un dossier de demande d’intervention de l’assurance obligatoire pour un renouvellement au Collège des médecins-directeurs sur base des formulaires B-Form-I-07 et 08. Ce dossier comprendra les documents de la première implantation démontrant que cette implantation répondait aux critères d’intervention de l’assurance obligatoire ainsi qu’un rapport médical de l’évolution, dans lequel doit être entre autres mentionné le tableau clinique depuis l’implantation et une comparaison avec le tableau clinique avant implantation ainsi que la justification du renouvellement.
Le Collège prendra la décision d’une intervention de l’assurance obligatoire pour le renouvellement, éventuellement sur base de l’avis de la Commission Peer Review.
7. Règles d’attestation
7.1 Règles de cumul et de non-cumul
Une intervention de l'assurance obligatoire pour la prestation 171496-171500, 171555-171566 ou 171614-171625 exclut, pendant une période de deux ans, une intervention de l'assurance pour la prestation 170892-170903.
7.2. Autres règles
Les prestations 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 et 171813-171824 suivent les modalités de remboursement de la catégorie A.
Les prestations 171614-171625, 171636-171640 ou 171651-171662 suivent les modalités de remboursement de la catégorie F.
L’intervention de l’assurance obligatoire pour la prestation 171511-171522 ne peut être accordée que minimum deux ans après la prestation 171496-171500, 171511-171522 ou 171533-171544.
L’intervention de l’assurance obligatoire pour la prestation 171570-171581 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 ou 171651-171662.
L’intervention de l’assurance obligatoire pour la prestation 171636-171640 ne peut être accordée que minimum neuf ans après la prestation 171555-171566, 171570-171581, 171592-171603,171614-171625, 171636-171640 ou 171651-171662..
7.3 Dérogation aux règles d’attestation
Pas d’application.
8. Engagements de l’établissement hospitalier
8.1. L’établissement hospitalier qui a adhéré à la convention est tenu de tenir à jour consciencieusement les caractéristiques de référence (baseline) et les données de suivi des bénéficiaires traités dans le cadre de cette convention afin de réaliser l'analyse prévue au point 9.2. Ces données doivent être conservées dans le Dossier Patient Informatisé.
8.2. Le pouvoir organisateur de l'établissement hospitalier porte la responsabilité de communiquer, sans délai, chaque modification au Fonctionnaire dirigeant du Service des soins de santé de l'INAMI et bien entendu à tous ceux qui sont concernés, dont en premier lieu les bénéficiaires.
Par e-mail au Secrétariat de la Commission de remboursement des implants et des dispositifs médicaux invasifs, à l’adresse suivante : implant@riziv-inami.fgov.be.
Lorsque le Fonctionnaire dirigeant constate des manquements, il informe les organismes assureurs du fait que le dispositif n’est plus remboursable pour cet établissement hospitalier.
9. Analyse
9.1. L’analyse des données émanant de cette application clinique limitée est effectuée par la Commission Peer Review et les organismes assureurs – nommés également évaluateur – qui ont signé la convention. Ils apportent un support technique et scientifique et évaluent les résultats du dispositif médical selon des critères précis.
9.2. Analyse – Rapport final
Au plus tard 6 mois avant la fin de la convention, l’évaluateur doit rédiger un rapport final sur base des données collectées et le transmettre à la Commission.
Ce rapport concerne tous les bénéficiaires implantés avec un neurostimulateur pour stimulation cérébrale profonde en cas d’épilepsie réfractaire depuis la date d’entrée en vigueur du remboursement.
Ce rapport final comprend au minimum les éléments suivants :
1) le nombre de patients traités, sexe, âge ;
2) analyse de tous les critères d’inclusion et d’exclusion dont indications, contre-indications, durée des symptômes, traitements, autres affections psychiatriques comorbides ;
3) résultats de la neurostimulation :
a. modification de la sévérité des crises
b. modification de la fréquence des crises
c. taux de répondeurs (= diminution de 50% ou plus de la fréquence des crises)
d. modification du traitement médicamenteux
4) effets secondaires, complications
5) une analyse rétrospective des coûts médicaux directs réalisée avec une comparaison des coûts cumulatifs pendant une période de un an avant l’implantation du stimulateur DBS avec les coûts cumulatifs de l’année suivant l’implantation. Les coûts étudiés seront les suivants :
a. nombre et durée des admissions en établissement hospitalier et liées à l’épilepsie. Les examens techniques relatifs au diagnostic et au traitement sont inclus, les coûts
dus à l’évaluation préchirurgicale ne font pas l’objet de l’étude
b. nombre de visite aux urgences
c. nombre de visite chez le médecin généraliste ou le neurologue
d. nombre et dose de spécialités pharmaceutiques antiépileptiques
6) une analyse des coûts indirects
7) une comparaison des résultats avec la littérature existante
Les organismes assureurs fourniront l’analyse rétrospective des coûts médicaux directs et indirects comme défini aux points 5 et 6 du contenu du rapport final.
Si ce rapport n’est pas communiqué à la date mentionnée ci-dessus, la Commission en informe le Ministre. Celui-ci peut prendre la décision de suspendre le remboursement du dispositif.
La Commission utilisera le rapport final qui évalue le dispositif comme base pour rédiger un règlement définitif. Ce règlement sera soumis au Ministre par l’intermédiaire de la Commission.
10. Droit de résiliation pour chaque partie prenante
La convention entre en vigueur le 1er décembre 2020 et est valable jusqu’au 30 novembre 2023 inclus mais peut toujours être résiliée par l’INAMI ou par un établissement hospitalier par lettre recommandée à la poste, adressée à l'autre partie, en respectant le délai de résiliation de trois mois qui prend cours le premier jour du mois suivant la date d'envoi de la lettre recommandée.
La convention expire dès que l’établissement hospitalier ne répond plus aux dispositions de cette convention.
11. Divers
A la demande de la Commission ou de l’évaluateur, une réunion peut être organisée à tout moment.
Teneinde een tijdelijke tegemoetkoming van de verplichte verzekering te kunnen genieten voor de verstrekkingen betreffende de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie moet aan volgende voorwaarden worden voldaan:
1. Doel van de overeenkomst
Deze overeenkomst heeft tot doel de tijdelijke tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging inzake de neurostimulatoren en hun toebehoren voor diepe hersenstimulatie in geval van refractaire epilepsie alsook de modaliteiten ervan te bepalen in het kader van een beperkte klinische toepassing gedurende de evaluatieperiode die loopt van 1 december 2020 tot en met 30 november 2023. Gedurende die periode wordt het hulpmiddel geëvalueerd volgens de bepalingen voorzien in punt 9.
2. Criteria betreffende de verplegingsinrichting
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien ze zijn uitgevoerd in een verplegingsinrichting die aan de volgende criteria voldoet en die de overeenkomst B-BKT-001-bis heeft afgesloten met het Verzekeringscomité:
De verplegingsinrichting moet gedurende de volledige looptijd van de overeenkomst voldoen aan de onderstaande criteria.
2.1. Enkel de verplegingsinrichtingen die de revalidatieovereenkomst van de referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden, hebben ondertekend, kunnen toetreden tot deze overeenkomst.
De verplegingsinrichting moet 24 uur op 24 en 7 dagen op 7 een neurochirurgische en neurologische permanentie hebben.
De indicatiestelling, de prechirurgische evaluatie, de implantatie, met inbegrip van de vervangingen, de revalidatie en een follow-up op lange termijn mogen enkel door deze verplegingsinrichtingen worden uitgevoerd. Het Comité van de verzekering voor geneeskundige verzorging stelt, op voorstel van de Dienst voor geneeskundige verzorging, een lijst op die continu wordt bijgewerkt, met vermelding van de samenstelling van het team per verplegingsinrichting en bezorgt die lijst ter informatie aan de Commissie.
De verplegingsinrichting en de artsen toegetreden tot de overeenkomst B-BKT-001-bis engageren zich tot het meewerken aan de evaluatie zoals bedoeld in punt 9.
De dagelijkse follow-up van de rechthebbende mag buiten die referentiecentra worden verricht.
2.2. Kandidatuurformulier voor de verplegingsinrichting
De verplegingsinrichting moet zich kenbaar maken bij de Dienst voor Geneeskundige verzorging op basis van het formulier B-Form-II-02.
2.3. Samenwerkingsakkoord
De verplegingsinrichting mag geen geformaliseerd samenwerkingsakkoord met andere verplegingsinrichtingen of met andere ziekenhuisverenigingen sluiten. Bovendien moet de ingreep binnen de muren van het bevoegde verplegingsinrichting worden uitgevoerd.
3. Criteria betreffende de rechthebbende
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640, 171651-171662, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien de rechthebbende aan de volgende criteria voldoet:
3.1. Inclusiecriteria
1) De rechthebbende lijdt aan focale epilepsie met focale complexe aanvallen met of zonder secundaire generalisering.
2) De rechthebbende lijdt aan refractaire epilepsie, d.w.z. waarbij de aanvallen onvoldoende onder controle kunnen worden gehouden met één van de potentieel doeltreffende anti-epileptica, die alleen of in combinatie worden toegediend, met optimale therapeutische dosissen waaraan geen onaanvaardbare bijwerkingen zijn verbonden; de aanvallen kunnen bovendien arbeidsongeschiktheid of een handicap tot gevolg hebben.
3) De rechthebbende moet een optimale farmacologische behandeling hebben gekregen. De rechthebbende is niet in staat om de epilepsie voldoende onder controle te houden met minstens 3 verschillende therapieën, waarvan minstens één combinatie, met optimale dosissen en gedurende een periode die volstaat om de doeltreffendheid ervan te beoordelen.
4) De rechthebbende komt niet in aanmerking voor chirurgie of de epilepsiechirurgie is een mislukking. De prechirurgische evaluatie omvat de volgende tests:
a. langdurige EEG-videomonitoring met registratie van de aanvallen
b. hogeresolutie-MRI van de hersenen
c. FDG-PET-scan van de hersenen
d. neuropsychologische evaluatie die de volgende elementen bevat:
i. IQ
ii. geheugenfuncties
iii.
e. psychiatrische evaluatie die onder andere de volgende elementen bevat:
i. Beck Depression Inventory
ii. QOLIE-31
In geval dit onderzoek niet gerealiseerd kan worden, bijvoorbeeld als gevolg van een mentale achterstand (die op zich geen tegenindicatie vormt voor de implantatie van een stimulator voor diepe hersenstimulatie), dient de reden duidelijk te worden vermeld op het formulier.
5) De rechthebbende moet op het moment van de implantatie minstens 18 jaar en maximaal 65 jaar oud zijn.
6) De rechthebbende kan een patiëntenprogrameerapparaat gebruiken en is bereid om zich aan de gevraagde tests te onderwerpen.
7) Alleen de rechthebbenden die duidelijk in staat zijn om, uit eigen wil, via een informed consent over de implantatie van elektroden en een neurostimulator te beslissen, komen in aanmerking. In die verklaring moeten omstandig de voor- en nadelen van de behandeling, de risico’s en de psychosociale impact worden toegelicht.
8) Als de rechthebbende met nervus vagusstimulatie wordt behandeld en indien daarmee onvoldoende resultaten worden bereikt, moet de rechthebbende vanaf de primo-implantatie minstens 2 jaar op de implantatie van een DBS-neurostimulator wachten en vice versa.
9) De gestimuleerde zone (doelgebied) is de zone die in de indicaties wordt vermeld en die door de huidige CE-markering wordt gedekt.
3.2. Exclusiecriteria
1) Ernstige neurologische of medische aandoening die een contra-indicatie is voor een cerebrale ingreep.
2) Elke chirurgische contra-indicatie om DBS te ondergaan, met inbegrip van de contra-indicaties die voor de DBS en/of voor de uitvoering van een preoperatieve MRI bekend zijn, contra-indicaties in het kader van een ingreep onder anesthesie of andere risicofactoraren voor een chirurgische ingreep (een ernstige cardiovasculaire aandoening, coagulopathie, …).
3) Ongepast gebruik van een product (alcohol, drugs, …)/middelenmisbruik waardoor het toestel niet correct kan worden gebruikt of waardoor er geen systematische medische/psychiatrische follow-up mogelijk is.
4) Suïcidale gedachten
5) Chronische psychotische problematiek die niet gestabiliseerd is onder behandeling, met uitzondering van een peri-ictale psychose
4. Criteria betreffende het hulpmiddel
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 kunnen enkel in aanmerking komen voor een tegemoetkoming van de verplichte verzekering indien het hulpmiddel aan de volgende criteria voldoet:
4.1. Definitie
De hulpmiddelen bedoeld onder de verstrekkingen 171614-171625, 171636-171640 en 171651-171662 dragen geen CE-markering, maar moeten het voorwerp uitmaken van een derogatie toegekend door de Minister die bevoegd is voor Volksgezondheid.
4.2. Criteria
Om te kunnen worden opgenomen op de nominatieve lijst voor de 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684 en 171695-171706 moet het hulpmiddel beantwoorden aan de volgende criteria:
De werkzaamheid en veiligheid van het hulpmiddel moeten zijn aangetoond met behulp van klinische studies. Deze studies dienen gepubliceerd te zijn in een internationaal erkend peer reviewed tijdschrift. Onder de gerealiseerde klinische studies, moet er minstens een follow-up studie zijn met minimum 100 patiënten die gedurende minimaal 2 jaar gevolgd worden, evenals een vergelijkende, gerandomiseerde klinische studie gedurende minimum 3 maand en met een a priori vastgelegde statistische power van minstens 80%. De follow-up studie en de gerandomiseerde studie mogen dezelfde studie zijn.
Een hulpmiddel dat een aanpassing is van een hulpmiddel dat reeds op de nominatieve lijst voor dezelfde verdeler ingeschreven is, zonder wijziging van het werkingsmechanisme en zonder negatieve impact op de werkzaamheid, de veiligheid en de kwaliteit, kan ingeschreven worden zonder klinische studies op voorwaarde dat de verdeler de aanpassingen en hun praktische consequenties beschrijft.
4.3. Garantievoorwaarden
Om te kunnen worden opgenomen op de nominatieve lijst voor de verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171791-171802 en 171813-171824 moet het hulpmiddel beantwoorden aan de volgende garantievoorwaarden:
- Niet heroplaadbare neurostimulatoren:
een totale garantie van 24 maanden moet worden gegeven voor de niet heroplaadbare neurostimulatoren
- Heroplaadbare neurostimulatoren:
een garantie van negen jaar moet worden gegeven voor de heroplaadbare neurostimulatoren: een volledige garantie voor de eerste vijf jaar en voor de volgende vier jaar een garantie pro rata. Voor de lader 171791-171802 en 171813-171824 is een volledige garantie van negen jaar vereist. De verstrekkingen 171614-171625, 171636-171640 en 171651-171662, moeten ook aan de garantievoorwaarden voldoen.
5. Aantal rechthebbenden
Het aantal rechthebbenden dat voor een tegemoetkoming van de verplichte verzekering in aanmerking kan komen, wordt beperkt tot 25 nieuwe rechthebbenden per jaar.
Zodra het aantal dreigt te worden overschreden, brengt het Secretariaat de Commissie op de hoogte en wordt aan de evaluator gevraagd een verslag te bezorgen. De aard van het verslag wordt vastgelegd door de Commissie.
De Commissie brengt de verplegingsinrichtingen en de verstrekkers van het betrokken hulpmiddel op de hoogte en neemt de noodzakelijke maatregelen.
6. Aanvraagprocedure en formulieren
6.1. Eerste implantatie
De aanvraag tot tegemoetkoming van de verplichte verzekering voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 gebeurt als volgt:
De terugbetalingsaanvraag voor de verstrekkingen 171496-171500, 171555-171566, 171614-171625, 171673-171684, 171710-171721, 171754-171765 en 171791-171802 wordt vóór de implantatie op basis van formulier B-Form-I-07 ingediend door de epileptoloog aan het College van artsen-directeurs.
Die verstrekkingen mogen enkel na akkoord van het College van artsen-directeurs door de verplichte verzekering worden terugbetaald. In geval van twijfel legt het College van artsen-directeurs de aanvraag voor advies voor aan de Commissie Peer Review, die bestaat uit zorgverleners die de revalidatieovereenkomst “referentiecentra voor rechthebbenden die aan refractaire epilepsie lijden” hebben ondertekend. Die Commissie Peer Review moet de aanvraag van het team van het referentiecentrum binnen de drie maanden onderzoeken. De Commissie Peer Review informeert het team van het referentiecentrum dat de aanvraag heeft ingediend, zodat deze de aanvraag kan verdedigen. Tijdens de bespreking van de dossiers die in aanmerking komen voor een tegemoetkoming van de verplichte verzekering voor neurostimulatie kan er altijd een lid van het College van artsen-directeurs of een arts, lid van de Commissie, aanwezig zijn. Minstens twee leden van de Commissie Peer Review die tot twee andere verplegingsinrichtingen dan het aanvragend referentiecentrum behoren, moeten hun akkoord geven.
De Commissie Peer Review bezorgt haar geargumenteerde conclusies (akkoord – weigering – uitstel) aan het College van artsen-directeurs.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging, eventueel op basis van het advies van de Commissie Peer Review, en dit voor elke rechthebbende afzonderlijk.
Het College van artsen-directeurs deelt zijn gemotiveerde beslissing mee binnen de dertig werkdagen. Als dit College een advies van de Commissie Peer Review heeft gevraagd, deelt het zijn gemotiveerde beslissing mee binnen de dertig werkdagen na ontvangst van het verslag van de Commissie Peer Review. Die beslissing wordt aan de epileptoloog van het betrokken referentiecentrum dat tot de overeenkomst is toegetreden, aan de betrokken rechthebbende via zijn verzekeringsinstelling en aan de ziekenhuisapotheker meegedeeld.
De documenten, waaruit blijkt dat voldaan is aan één van de indicaties vermeld onder punt 3, moeten steeds in het medisch dossier van de rechthebbende aanwezig zijn.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171614-171625 worden geattesteerd.
6.2. Vervanging
In geval van hernieuwing van een hulpmiddel dat reeds het voorwerp heeft uitgemaakt van een tegemoetkoming van de verplichte verzekering in het kader van deze overeenkomst, mag de aanvraag voor de verstrekkingen 171511-171522, 171570-171581 171636-171640, 171695-171706, 171732-171743, 171776-171780 en 171813-171824 na implantatie ingediend worden door de epileptoloog bij het College van artsen-directeurs door middel van het aanvraagformulier B-Form-I-08.
De aanvraag bevat onder andere een medisch evolutieverslag, waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de redenen voor de noodzaak van de hernieuwing zijn opgenomen.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171636-171640 worden geattesteerd.
6.3. Voortijdige vervanging
De procedure beschreven onder punt 6.2. dient te worden toegepast.
Tot de verkrijging van een CE-markering mogen de heroplaadbare neurostimulatoren enkel worden geïmplanteerd nadat de Minister die bevoegd is voor Volksgezondheid daartoe een derogatie heeft toegekend. Een kopie van die derogatie zal aan het College van artsen-directeurs worden bezorgd.
In dit geval moet de verstrekking 171651-171662 worden geattesteerd.
6.4. Derogatie aan de procedure
Voor de rechthebbenden die reeds vóór de inwerkingtreding van deze overeenkomst ingeplant zijn en die vóór de implantatie aan alle voorwaarden bedoeld in punt 3 voldeden, kan een tegemoetkoming van de verplichte verzekering voor de hernieuwing van de neurostimulatoren en toebehoren worden toegekend volgens de modaliteiten voorzien in punt 6.1.
In dat geval bezorgt het team van het referentiecentrum een aanvraagdossier tot tegemoetkoming van de verplichte verzekering voor een hernieuwing aan het College van artsen-directeurs op basis van de formulieren B-Form-I-07 en 08. Dat dossier bevat de documenten van de eerste implantatie waarin wordt aangetoond dat deze implantatie aan de vergoedingscriteria van de verplichte verzekering voldeed en een medisch evolutieverslag waarin onder meer het klinisch beeld sinds de implantatie, een vergelijking met het klinisch beeld vóór de implantatie en de rechtvaardiging van de hernieuwing zijn opgenomen.
Het College van artsen-directeurs neemt de beslissing voor een tegemoetkoming van de verplichte verzekering voor de hernieuwing, eventueel op basis van het advies van de Commissie Peer Review.
7. Regels voor attestering
7.1. Cumul en non-cumulregels
Een tegemoetkoming van de verplichte verzekering voor de verstrekking 171496-171500, 171555-171566 of 171614-171625 sluit tijdens een periode van twee jaar een tegemoetkoming van de verzekering voor de verstrekking 170892-170903 uit.
7.2. Andere regels
De verstrekkingen 171496-171500, 171511-171522, 171533-171544, 171555-171566, 171570-171581, 171592-171603, 171673-171684, 171695-171706, 171710-171721, 171732-171743, 171754-171765, 171776-171780, 171791-171802 en 171813-171824 volgen de vergoedingsmodaliteiten van categorie A.
De verstrekkingen 171614-171625, 171636-171640 en 171651-171662 volgen de vergoedingsmodaliteiten van categorie F.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171511-171522 kan pas minstens twee jaar na de verstrekking 171496-171500, 171511-171522 of 171533-171544 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171570-171581 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171614-171625, 171636-171640 of 171651-171662 worden toegekend.
De tegemoetkoming van de verplichte verzekering voor de verstrekking 171695-171706 kan pas minstens negen jaar na de verstrekking 171555-171566, 171570-171581, 171592-171603, 171625, 171636-171640 of 171651-171662 worden toegekend.
7.3. Derogatie van de attesteringsregels
Niet van toepassing.
8. Verbintenissen van de verplegingsinrichting
8.1. De verplegingsinrichting die tot de overeenkomst is toegetreden moet de baseline karakteristieken en de follow-up gegevens van de rechthebbenden die in het kader van deze overeenkomst zijn behandeld nauwgezet bijhouden om de analyse voorzien in punt 9.2. uit te voeren. Deze gegevens moeten in het Elektronisch Patiënten Dossier bewaard worden.
8.2. De inrichtende macht van de verplegingsinrichting is verantwoordelijk voor de onverwijlde mededeling van elke wijziging aan de leidend ambtenaar van de Dienst voor geneeskundige verzorging van het RIZIV en natuurlijk aan alle betrokkenen, waaronder in de eerste plaats de rechthebbenden.
Per e-mail aan het Secretariaat van de Commissie Tegemoetkoming Implantaten en Invasieve Medische Hulpmiddelen, op het volgende adres: implant@riziv-inami.fgov.be.
Wanneer de Leidend ambtenaar nalatigheden vaststelt, brengt hij de verzekeringsinstellingen ervan op de hoogte dat het hulpmiddel voor die verplegingsinrichting niet meer mag worden terugbetaald.
9. Analyse
9.1. De analyse van de gegevens uit deze beperkte klinische toepassing wordt uitgevoerd door de Commissie Peer Review en de verzekeringsinstellingen – verder evaluator genoemd – die mee de overeenkomst ondertekend hebben. Zij verlenen technische en wetenschappelijke ondersteuning en analyseren de resultaten van het medisch hulpmiddel volgens precieze criteria.
9.2. Analyse – Eindverslag
Uiterlijk 6 maanden voor het einde van de overeenkomst moet de evaluator op basis van de verzamelde gegevens een eindverslag opstellen en aan de Commissie bezorgen.
Dit verslag betreft alle rechthebbenden geïmplanteerd met een neurostimulator voor diepe hersenstimulatie in geval van refractaire epilepsie sinds de datum van inwerkingtreding van de terugbetaling.
Dat eindverslag moet minstens de volgende elementen bevatten:
1) het aantal behandelde patiënten, geslacht, leeftijd;
2) analyse van alle inclusie- en exclusiecriteria, waaronder indicaties, contra-indicaties, duur van de symptomen, behandelingen, andere comorbide psychiatrische aandoeningen;
3) resultaten van de neurostimulatie:
a. wijziging van de ernst van de aanvallen
b. wijziging van de aanvalsfrequentie
c. responsratio (= daling met 50 % of meer van de aanvalsfrequentie)
d. wijziging van de medicamenteuze behandeling
4) bijwerkingen, complicaties;
5) een retrospectieve analyse van de directe medische kosten, gerealiseerd met een vergelijking van de cumulatieve kosten gedurende één jaar vóór de implantatie van een DBS-stimulator met de cumulatieve kosten van het jaar na implantatie. De volgende kosten worden bestudeerd:
a. aantal en duur van opnames in verplegingsinrichting en gelinkt aan epilepsie. De technische onderzoeken met betrekking tot diagnostiek of behandeling zijn inbegrepen.
De kosten te wijten aan de prechirurgische evaluatie zijn niet het onderwerp van de studie.
b. aantal bezoeken aan de spoed
c. aantal bezoeken bij de huisarts of de neuroloog
d. aantal en dosis van de anti-epileptische farmaceutische specialiteiten
6) een analyse van de indirecte kosten;
7) een vergelijking van de resultaten met de bestaande literatuur.
De verzekeringsinstellingen zullen de retrospectieve analyse van de directe en indirecte medische kosten zoals gedefinieerd in punten 5 en 6 van de inhoud van het eindverslag bezorgen.
Indien dit verslag niet op de voormelde datum wordt meegedeeld, brengt de Commissie de minister daarvan op de hoogte. Deze kan beslissen om de terugbetaling van het hulpmiddel stop te zetten.
De Commissie zal het eindverslag waarin het hulpmiddel wordt geëvalueerd, als basis kunnen gebruiken voor het opstellen van een definitieve regeling. Die regeling zal door de Commissie aan de Minister worden voorgelegd.
10. Opzeggingsrecht voor elke betrokken partij
De overeenkomst treedt in werking op 1 december 2020 en is geldig tot en met 30 november 2023 maar kan steeds door het RIZIV of door een verplegingsinrichting worden opgezegd met een ter post aangetekende brief die aan de andere partij wordt gericht, mits inachtneming van een opzeggingstermijn van drie maanden die ingaat op de eerste dag van de maand volgend op de datum van verzending van de aangetekende brief.
De overeenkomst verstrijkt zodra de verplegingsinrichting niet meer aan de bepalingen van deze overeenkomst voldoet.
11. Varia
Op verzoek van de Commissie of van de evaluator kan er op elk moment een vergadering worden georganiseerd.
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.
Vergoedingsvoorwaarde B-§09
Interpretatieregels 171496 - 171500
Algemene vergoedingsvoorwaarde GNL-01
1.1. Les prestations reprises
sous le point 2. Prestations et Modalités de remboursement ne sont remboursées
que si elles sont prescrites par un médecin spécialiste et si elles répondent
aux dispositions spécifiques de ces prestations.
1.2. Si dans une condition de
remboursement (1. Critères concernant l’établissement hospitalier), il est fait
référence aux années 2020, 2021 ou 2022, le nombre de prestations pour chacune
de ces années sera remplacé par le nombre de prestations pour l’année 2019
(correspondant à l’année qui précède l’année où l’arrêté royal n°21 du 14 mai
2020, portant des adaptations temporaires aux conditions de remboursement et
aux règles administratives en matière d'assurance obligatoire soins de santé
suite à la pandémie COVID-19, est entré en vigueur) pour autant que le nombre
de prestations pour l’année 2019 soit supérieur à celui des prestations pour
l’année à laquelle il est fait référence.
1.3. Les dispositifs repris au point « 2. Prestations et modalités de remboursement » peuvent bénéficier d'une intervention de l’assurance obligatoire après avoir subi une légère modification telle que définie à l'article 1er, 51° de l'arrêté royal modifiant l'arrêté royal du 25 juin 2014 fixant les procédures, délais et conditions en matière d’intervention de l’assurance obligatoire soins de santé et indemnités dans le coût des implants et des dispositifs médicaux invasifs, et après que ces dispositifs aient suivi avec succès la procédure prévue à cet effet telle que décrite à l'article 145, § 2 jusqu'à l'article 152 du même arrêté.
1.4. Le terme «matériel implantable» dans un
libellé d’une prestation en catégorie II (Dispositifs médicaux invasifs autres
que pour usage à long terme) de la Liste fait référence à un dispositif médical
implantable tel que défini par le règlement (UE) 2017/745 (MDR) utilisé lors
d’une procédure de viscérosynthèse ou endoscopique et servant à faire une
ligature ou une suture (y compris les renforts de suture), à l’exception des
dispositifs médicaux qui font l’objet d’une intervention de l’assurance via une
autre prestation spécifique de la Liste.
1.1. De verstrekkingen opgenomen onder punt 2.
Verstrekkingen en Vergoedingsmodaliteiten worden enkel vergoed indien ze door
een arts-specialist zijn voorgeschreven en beantwoorden aan de specifieke
bepalingen bij die verstrekkingen.
1.2. Indien er in een vergoedingsvoorwaarde (1.
Criteria betreffende de verplegingsinrichting) verwezen wordt naar de jaren
2020, 2021 of 2022, wordt het aantal verstrekkingen voor elk van deze jaren
vervangen door het aantal verstrekkingen voor het jaar 2019 (dat overeenstemt
met het jaar voorafgaand aan het jaar waarin het Koninklijk Besluit nr. 21 van
14 mei 2020, betreffende tijdelijke aanpassingen van de vergoedingsvoorwaarden
en administratieve voorschriften voor de verplichte ziekteverzekering naar
aanleiding van de COVID-19-pandemie, in werking is getreden) op voorwaarde dat
het aantal verstrekkingen voor het jaar 2019 hoger is dan het aantal
verstrekkingen voor het jaar waarnaar wordt verwezen.
1.3 De hulpmiddelen opgenomen onder punt “2. Verstrekkingen en Vergoedingsmodaliteiten” kunnen in aanmerking komen voor een tegemoetkoming van de verplichte verzekering na een lichte wijziging te hebben ondergaan zoals gedefinieerd in artikel 1, 51° van het koninklijk besluit tot wijziging van het koninklijk besluit van 25 juni 2014 tot vaststelling van de procedures, termijnen en voorwaarden inzake de tegemoetkoming van de verplichte verzekering voor geneeskundige verzorging en uitkeringen in de kosten van implantaten en invasieve medische hulpmiddelen, en nadat deze hulpmiddelen de hiervoor bestemde procedure zoals beschreven in artikel 145, § 2 t.e.m. artikel 152 van datzelfde besluit succesvol hebben doorlopen.
1.4. De formulering “implanteerbaar materiaal” in een omschrijving van een verstrekking in categorie II (Invasieve medische hulpmiddelen voor niet-langdurig gebruik) van de Lijst verwijst naar een implanteerbaar medisch hulpmiddel zoals bedoeld in de Verordening (UE) 2017/745 (MDR) gebruikt tijdens een viscerosynthese of endoscopische ingreep en gebruikt om een ligatuur of hechting te doen (hechtingsversterkingen inbegrepen), met uitzondering van de medische hulpmiddelen die via een andere verstrekking van de Lijst van een tegemoetkoming van de verzekering genieten.